Risperidone and olanzapine have been shown to be both well tolerated and efficacious in the treatment of psychotic disorders
(1–
6). Almost half of all new prescriptions for antipsychotics in the United States are for these two medications. Large separate trials performed to obtain regulatory approval for risperidone and olanzapine have compared these newer agents with haloperidol. The findings have suggested that both drugs may be superior to haloperidol in amelioration of negative symptoms and that risperidone is superior in amelioration of the positive symptoms of schizophrenia
(3,
5). Overall, second-generation drugs do provide clear advantages in terms of fewer adverse effects, particularly drug-induced parkinsonism, akathisia, and tardive dyskinesia. Advantages in alleviating refractory symptoms, negative symptoms, depression, and suicidal behavior have been reported; however, much remains to be done methodologically in establishing the relative merits of specific drugs in these multiple domains of interest
(6). Only now are actual head-to-head studies being conducted; these will contribute to establishing relative efficacy and safety in relevant study populations.
We report the results of a double-blind, randomized trial comparing the safety and efficacy of risperidone and olanzapine at clinically common doses in 377 people with a DSM-IV diagnosis of schizophrenia or schizoaffective disorder.
Method
Design
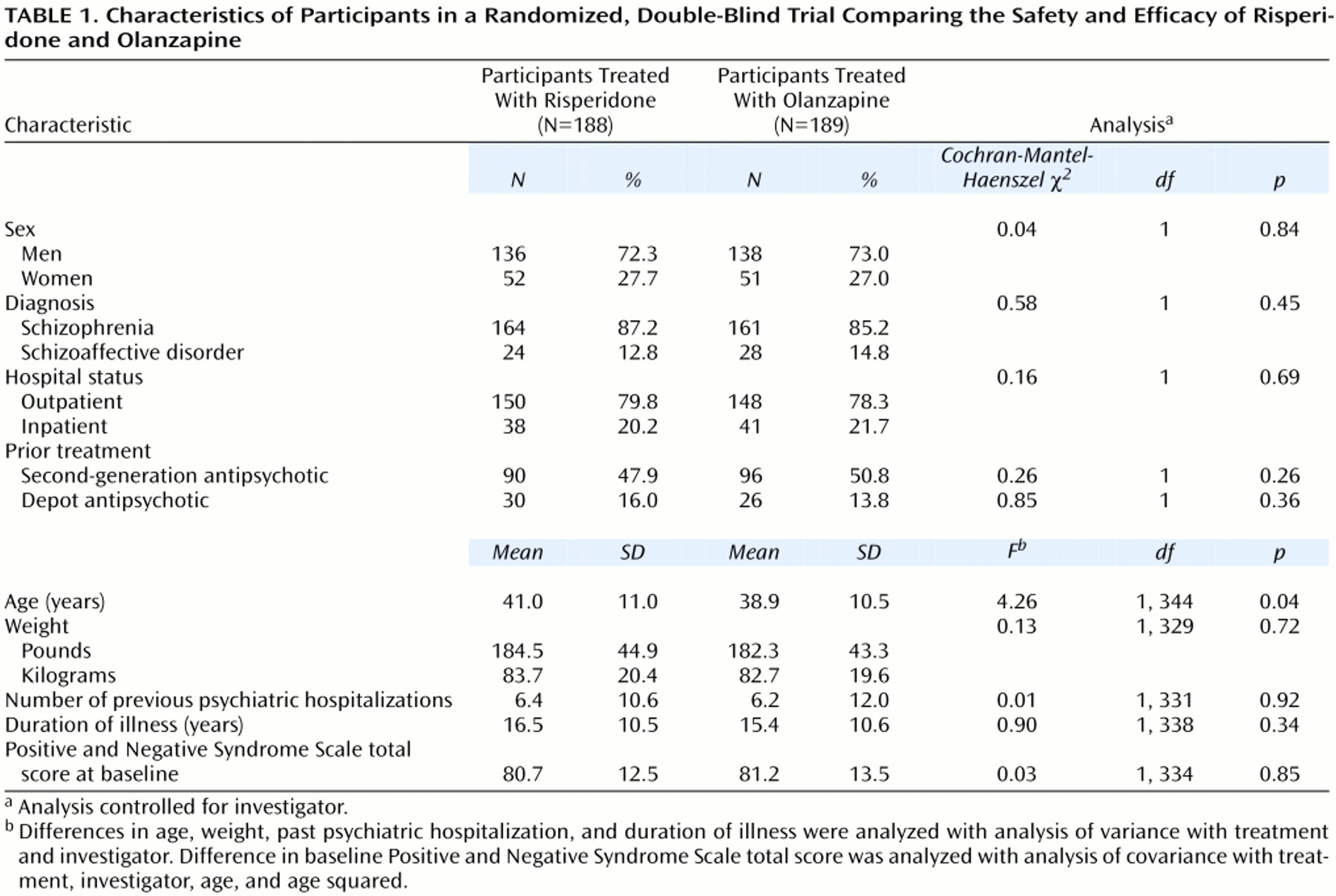
The trial was a multicenter, randomized, double-blind, parallel-group comparison of risperidone and olanzapine conducted at 41 sites in the United States. Two sites (30 subjects) were excluded from the analysis because of noncompliance with regulatory requirements. The quality of data from the remaining 39 sites was verified by thorough poststudy auditing of all 377 participant files.
After complete description of the study to the subjects, written informed consent was obtained. During the week before the subject’s assignment to one of the two treatment groups, all oral antipsychotic and anticholinergic medications currently taken by the subjects were gradually discontinued. Any depot antipsychotics were discontinued for at least one treatment cycle before the subject was assigned to a study group. The 377 participants were then randomly assigned to receive risperidone (2–6 mg/day) or olanzapine (5–20 mg/day) for 8 weeks.
Eligibility Criteria
Inclusion criteria included a DSM-IV diagnosis of schizophrenia or schizoaffective disorder, a baseline Positive and Negative Syndrome Scale
(12) score of 60 to 120 (calculated by using 1–7 scoring), and age 18–64 years. Participants could be outpatients or inpatients hospitalized ≤4 weeks at the time of screening. Informed consent was obtained from either the participant or a legal guardian or representative.
Exclusion criteria included a DSM-IV axis I diagnosis other than schizophrenia or schizoaffective disorder, a DSM-IV diagnosis of substance abuse in the 3 months before selection, documented disease of the central nervous system, the use of disallowed concomitant therapy such as mood stabilizers or antidepressants, a history of treatment with clozapine for more than 4 consecutive weeks, or being known by the investigator to be sensitive or unresponsive to risperidone or olanzapine.
Dosing Regimen
Both drugs were given once daily according to the following regimen: days 1–2, 2 mg/day of risperidone or 10 mg/day of olanzapine; days 3–7, 2–4 mg/day of risperidone or 5–10 mg/day of olanzapine; days 8–14, 2–6 mg/day of risperidone or 5–15 mg/day of olanzapine; and days 15–56, 2–6 mg/day of risperidone or 5–20 mg/day of olanzapine.
Assessments
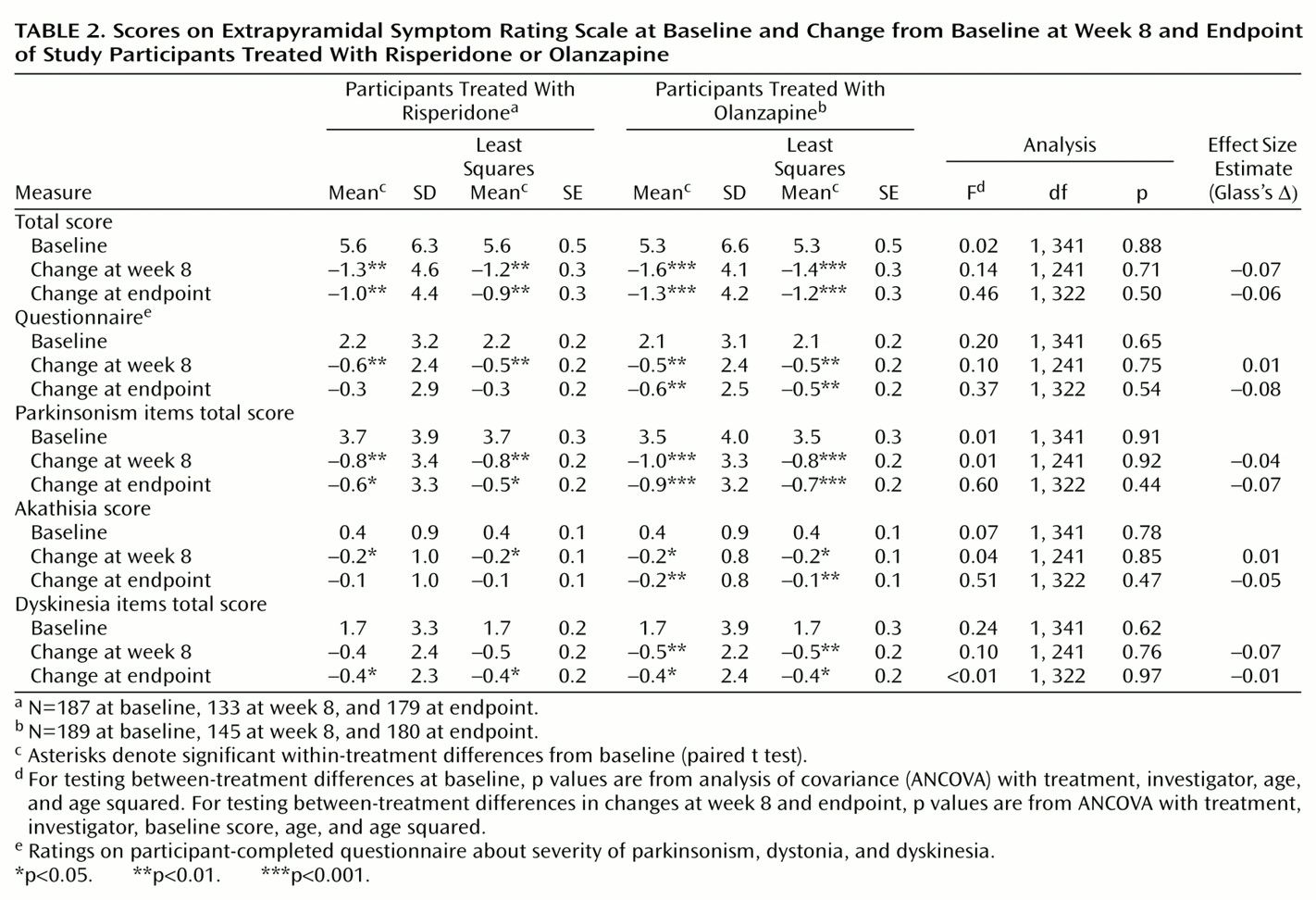
Participants were seen once a week. Participants’ reports of extrapyramidal symptoms were recorded at each study visit. Severity of extrapyramidal symptoms and psychopathology was evaluated with the Extrapyramidal Symptom Rating Scale
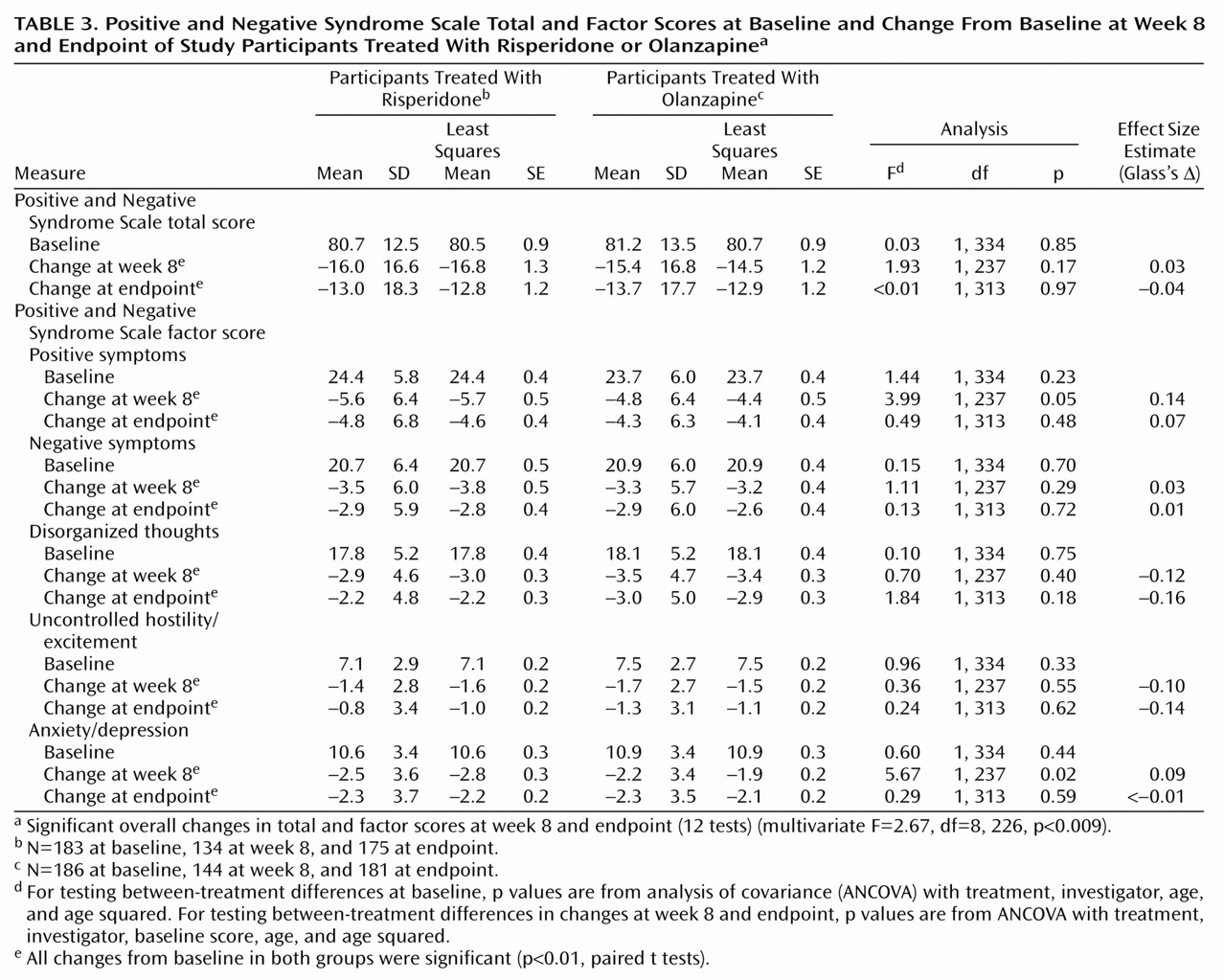
(13) and the Positive and Negative Syndrome Scale, respectively, at baseline and at weeks 2, 4, 6, and 8 (or withdrawal). Positive and Negative Syndrome Scale total scores and scores on five Positive and Negative Syndrome Scale factors
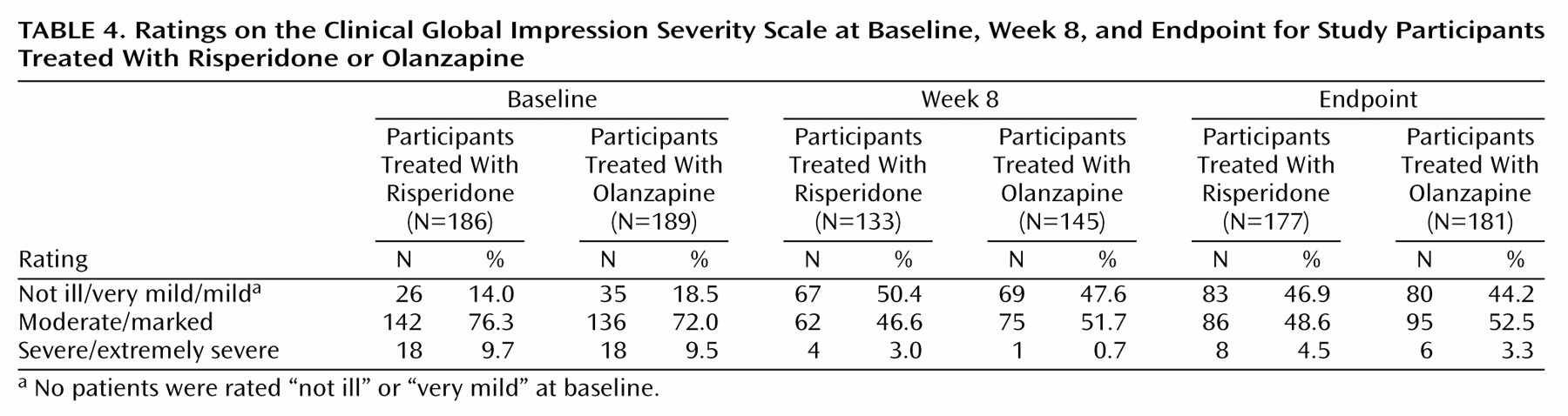
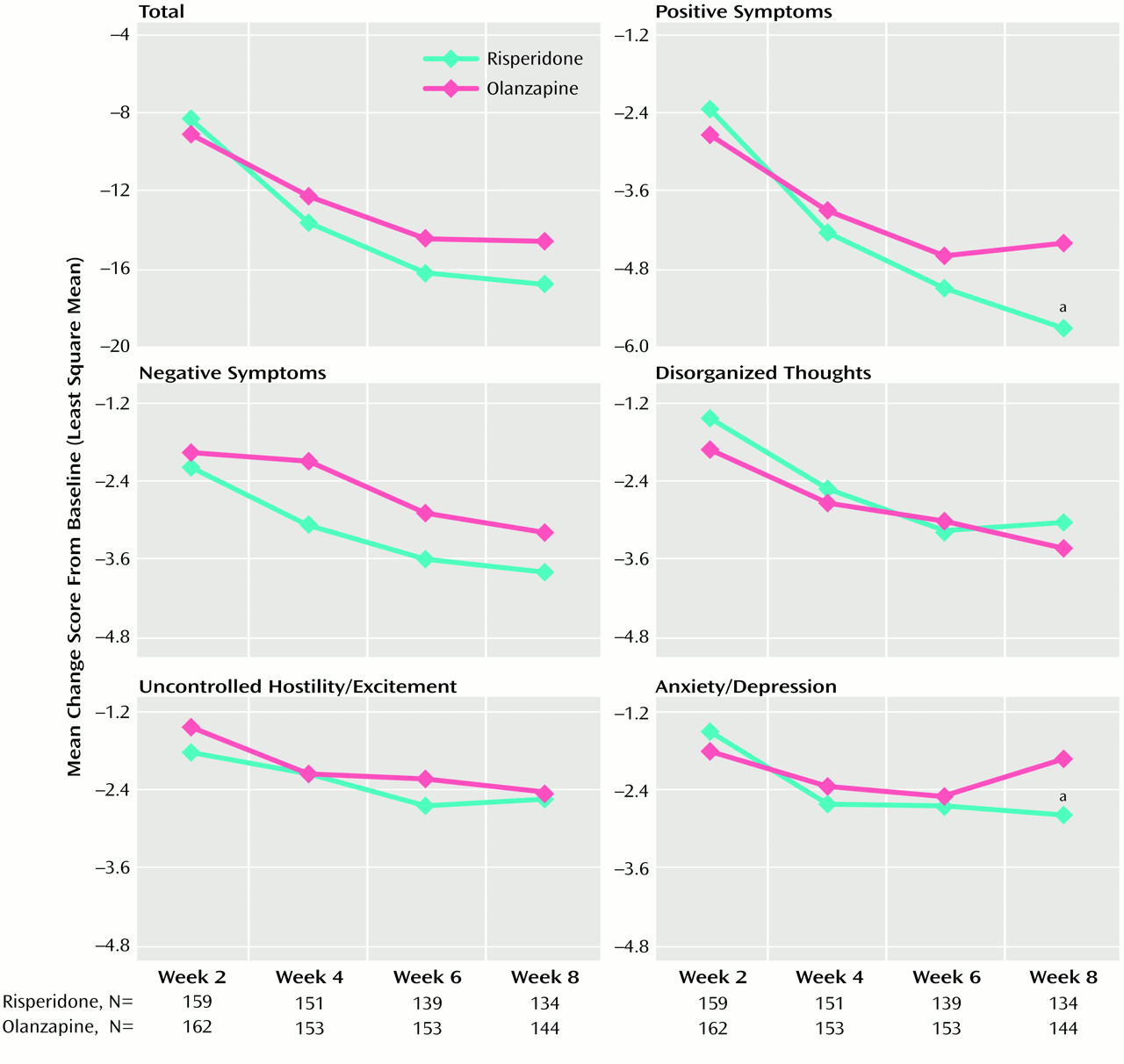
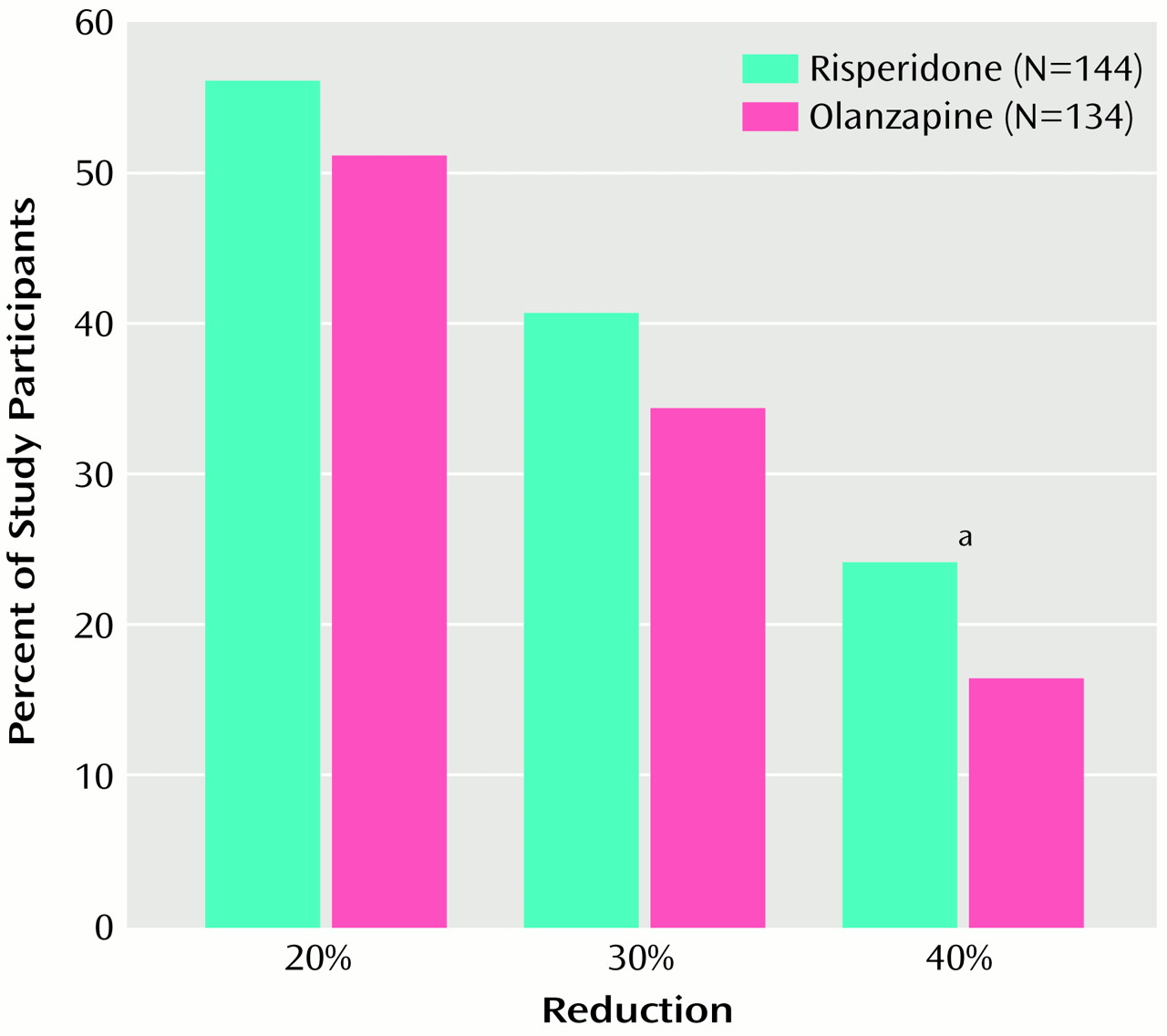
(3) were used to evaluate changes in symptom severity. The overall severity of illness was rated with the severity scale of the Clinical Global Impression (CGI) scale, and changes in severity were assessed with the CGI change scale. Clinical improvement was defined as a ≥20% reduction in total Positive and Negative Syndrome Scale scores.
Adverse events were recorded weekly. Standard laboratory tests were performed at randomization and weeks at 2, 4, 6, and 8 (or withdrawal). Vital signs were recorded at screening, randomization, and weeks 1, 2, 3, 4, 6, and 8. An ECG was performed at screening and at week 8 (or withdrawal). Participants completed self-assessment questionnaires at baseline and week 8 (or withdrawal), including a questionnaire that inquired about symptoms potentially related to prolactin in three areas (menstrual changes, breast or chest symptoms, and male sexual function).
Statistical Analysis
The size of the study group was determined on the basis of the difference in expected rates of spontaneously reported extrapyramidal symptoms (obtained from medication package inserts). Statistical tests on all data were performed at the 5%, two-tailed significance level. The principal analysis of tolerability and efficacy measures was conducted on an intent-to-treat basis, meaning that data for all participants who were treated and who had at least one postbaseline assessment were included in the analysis. Between-group comparisons of continuous measures were analyzed with analysis of covariance with the following factors: investigator, baseline values (Positive and Negative Syndrome Scale and Extrapyramidal Symptom Rating Scale), and age. To address issues of multiple comparisons in efficacy, a single multivariate F test was used to compare treatment groups on 12 Positive and Negative Syndrome Scale tests (total Positive and Negative Syndrome Scale score and scores on the five Positive and Negative Syndrome Scale factors at week 8 and at endpoint) with the following factors: treatment, investigator, age, age squared, and baseline scores. Within-group differences were then evaluated with paired t tests. Results at week 8 (observed-case analysis) and endpoint (missing data estimated by carrying the last observation forward) were considered primary. Overall differences in categoric measures were examined by using the Cochran-Mantel-Haenszel chi-square test controlling for investigator and, if necessary, baseline stratum. The ratio of beneficial to adverse changes in observed laboratory test values was computed by dividing the reduction from an above-normal score at baseline to a normal score by the increase from a normal score at baseline to an above-normal score. Between-treatment risk ratios were computed as the previous ratio for the risperidone group divided by the value for the olanzapine group. Because maximal drug exposure and thus potential toxicity occurred among the participants who remained in the trial for 8 weeks, 8-week results are reported. In general, as would be expected with a shorter period of drug exposure, endpoint analyses produced ratios closer to 1.
Discussion
The selection of an antipsychotic agent to treat people with schizophrenia or schizoaffective disorder is a complex decision for which the physician must weigh individual patient factors and numerous drug factors, including efficacy, safety, tolerability, and cost. In this large prospective study of risperidone and olanzapine in clinically relevant doses, both antipsychotic drugs were generally safe and efficacious in treating people with schizophrenia or schizoaffective disorder. Significant reductions in the severity of symptoms of schizophrenia and schizoaffective disorder, as measured by scores on the Positive and Negative Syndrome Scale, were seen at 2 weeks after initiation of treatment, and further improvements were noted throughout the 8-week trial.
The mean modal doses of risperidone (4.8 mg/day) and olanzapine (12.4 mg/day) received by the participants in this study are similar to those used in current clinical practice (4.5 mg/day of risperidone and 12.7 mg/day of olanzapine) (national dosing data for schizophrenia only)
(14). In contrast, in a previous comparative study of risperidone and olanzapine
(7), risperidone doses (mean modal dose=7.2 mg/day) were 60% higher than those selected for use in current clinical practice, as reflected by the same national dosing data. Optimal dosing is important to study interpretation. In addition to reducing tolerability, excessive doses may actually reduce efficacy, perhaps because of the loss of an optimal receptor occupancy (e.g., D
2) range or of an optimal binding pattern across different receptors (e.g., across 5-HT, D
2, and α
2)
(15,
16). Severity of extrapyramidal symptoms was reduced over the course of the study in both groups, with no significant between-group differences. The presence of extrapyramidal symptoms also may limit clinical response.
With respect to efficacy, a multivariate analysis of Positive and Negative Syndrome Scale scores revealed significant between-treatment differences. This may be attributable to the finding that on two Positive and Negative Syndrome Scale factors (positive symptoms and anxiety/depression) reductions in symptom severity were greater with risperidone than with olanzapine among participants who completed the 8-week study (p<0.05). In most comparisons of Positive and Negative Syndrome Scale factors, risperidone and olanzapine were equally efficacious. The finding that risperidone may have superior positive symptom efficacy compared with olanzapine is consistent with previously published research, including both an open direct-comparison study
(11) and large controlled trials
(3,
4) in which risperidone, but not olanzapine, was superior to haloperidol in reduction of positive symptoms. Potential superiority of risperidone in relief of mood symptoms in this population is an interesting finding, possibly related to large differences between the drugs in α
2 receptor affinity. Further research is needed to confirm this finding.
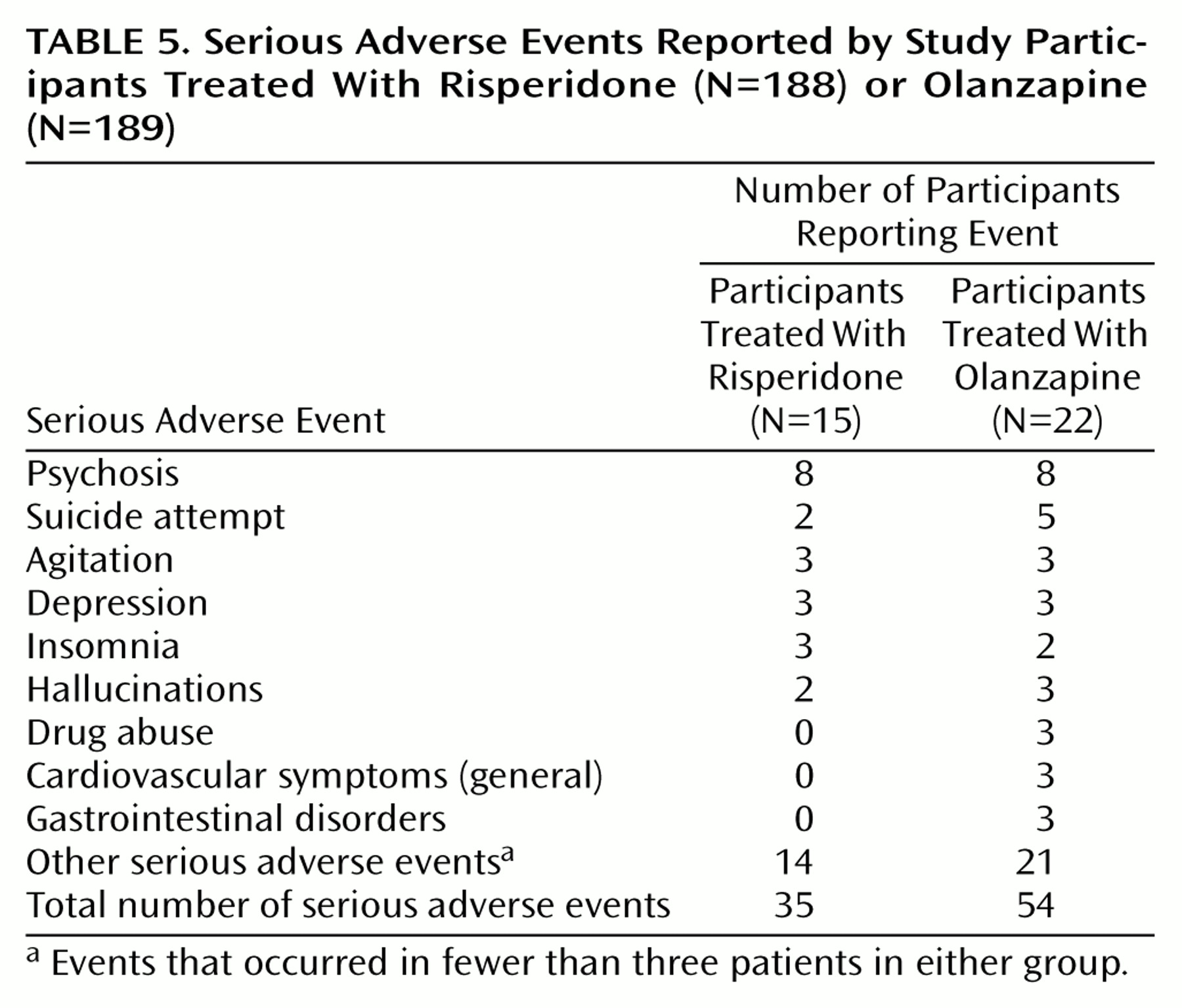
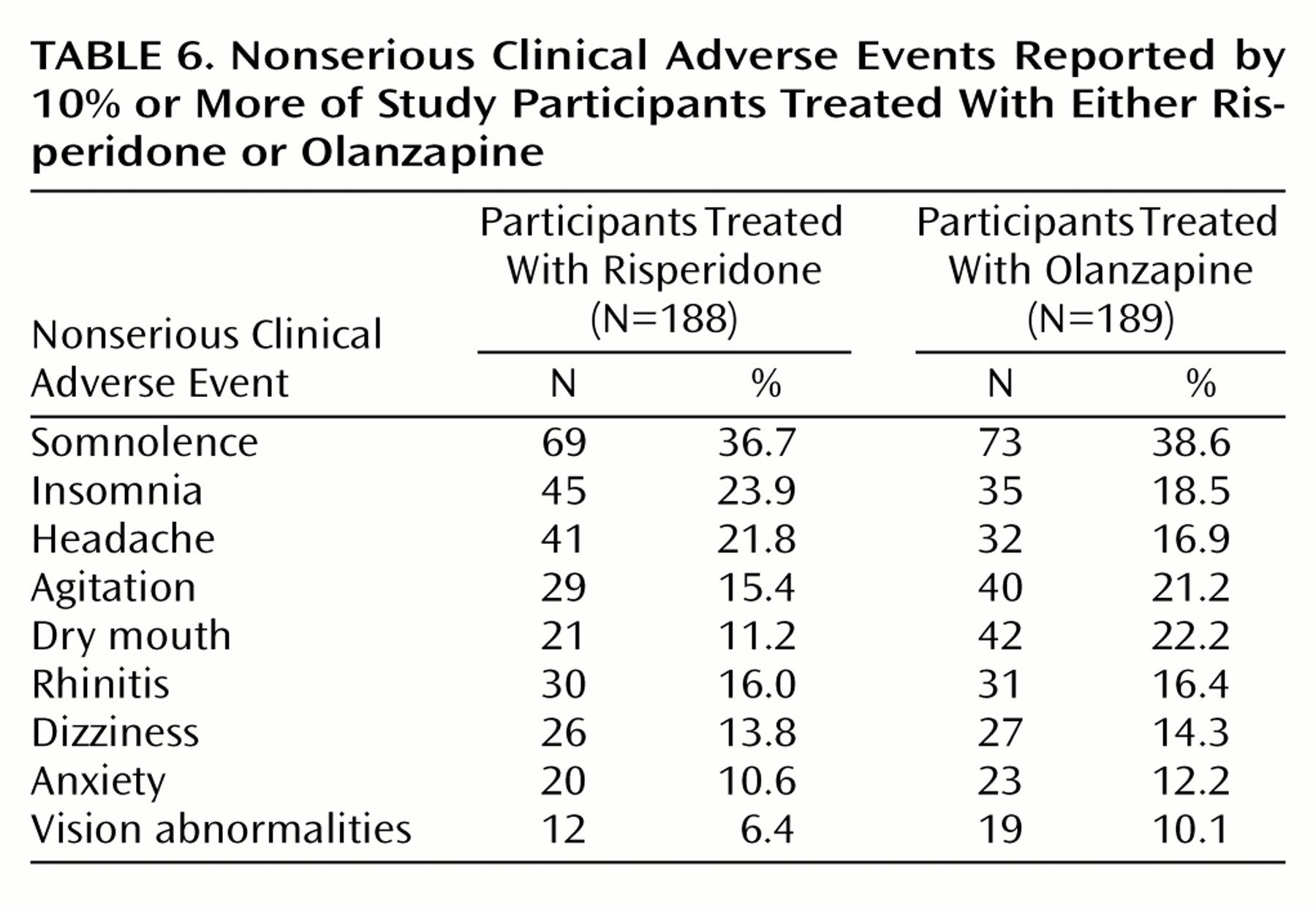
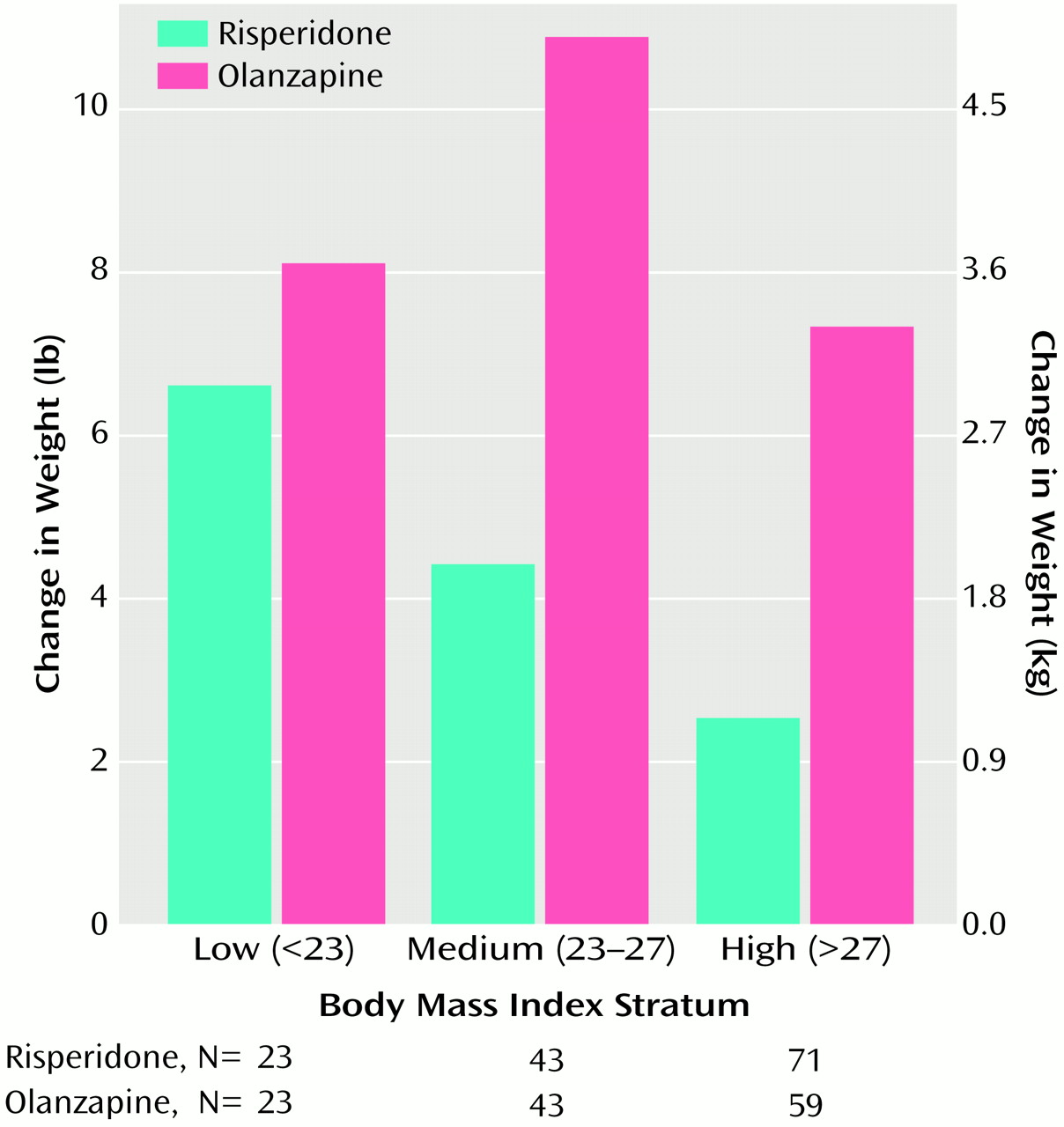
Few participants experienced serious adverse events, and somnolence was the only adverse event reported by 30% or more of the participants. There were greater increases in weight in participants treated with olanzapine (weight gain averaged about 1 lb per week in these participants) than in those treated with risperidone. Moreover, as shown in
Figure 4, weight gain with olanzapine was observed irrespective of participants’ baseline body mass index; in contrast, weight gain with risperidone was predominantly seen in participants with a low body mass index. More than 25% of the olanzapine participants (and 12% of risperidone participants) experienced a weight gain of 7% or more, an increase that is believed to pose health risks
(17). It is noteworthy that during this 8-week study some participants gained >20% of body weight (3% of olanzapine participants and no risperidone participants). Substantial health risks are associated with weight gain, a factor deserving careful consideration in long-term therapy. These health risks may include coronary heart disease, stroke, osteoarthritis, and breast, prostate, and colon cancers
(17), as well as possible adverse effects on compliance, self-esteem, and cost. Osser et al.
(18) have reported increases in weight and serum triglyceride levels with olanzapine. Wirshing et al.
(19), Goldstein et al.
(20), and Gatta et al.
(21) have reported new-onset diabetes and diabetic ketoacidosis in patients treated with olanzapine. Of particular concern may be the incremental risk with other risk factors common in this population (e.g., smoking)
(22). Factors that affect tolerability, such as extrapyramidal symptoms or liver transaminase or prolactin elevation, generally resolve rapidly with treatment modification, but weight gain may be persistent and refractory to treatment even after drug discontinuation. Substantial weight increases with olanzapine have been reported previously
(23,
24).
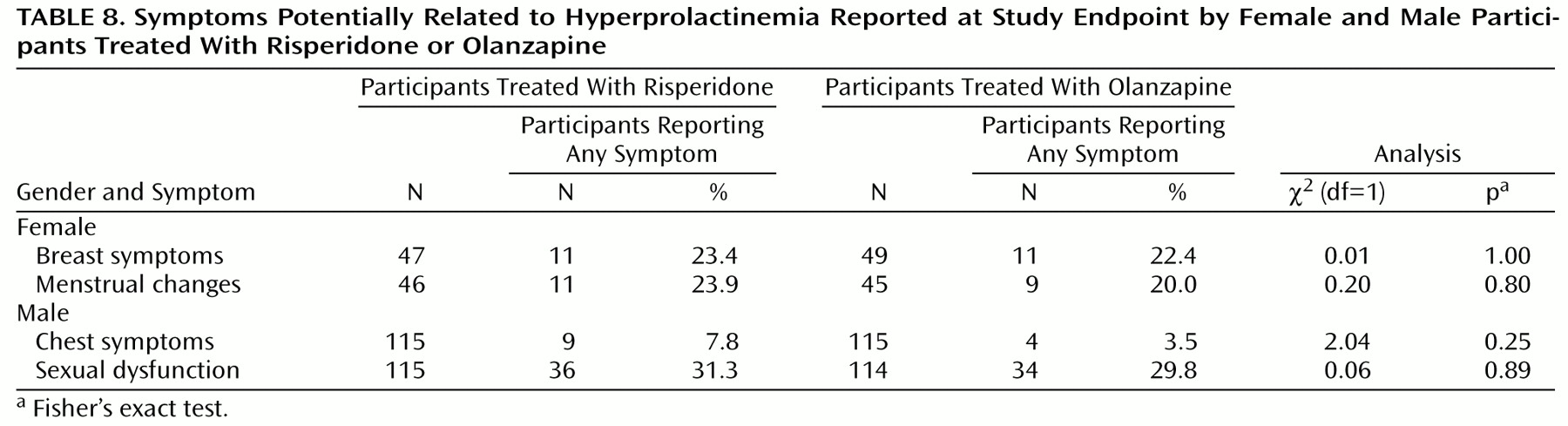
Prolactin levels were elevated in participants receiving risperidone. However, results of a questionnaire that solicited reports of symptoms potentially related to prolactin showed no difference between treatments. A similar lack of association between prolactin levels and adverse events potentially related to prolactin was found by Kleinberg et al.
(25) in their analysis of data from more than 800 men and women treated with risperidone. Although risperidone-induced elevated prolactin levels have been reported to produce clinical effects
(26–
28), we found such effects to be rare. This is not to say that symptoms are uncommon, merely that the association of such symptoms with prolactin levels may be the exception rather than the rule.
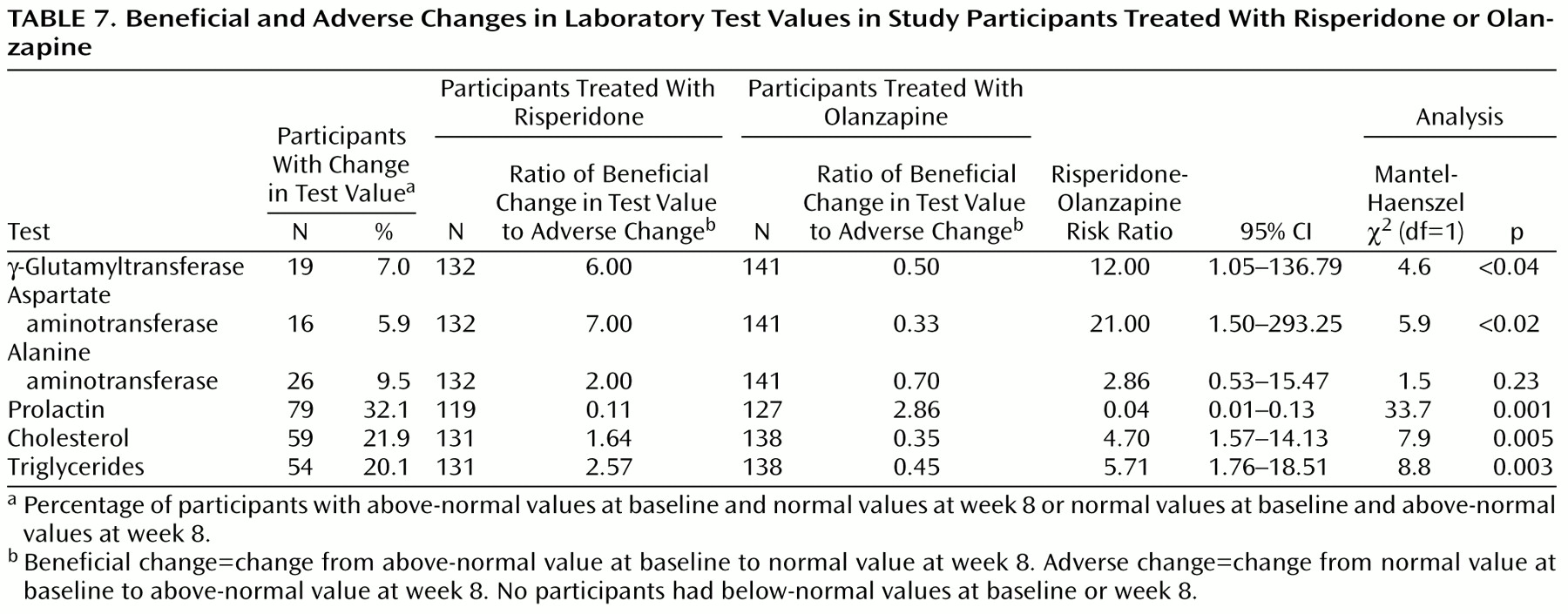
The ratio of beneficial-to-adverse change in laboratory test values (
Table 7) suggested potentially greater risk for undesirable changes in liver enzymes and serum lipid levels associated with olanzapine treatment, but as with the prolactin laboratory test values, these findings should be interpreted with caution in light of the low proportion of participants affected by such changes and the lack of related spontaneously reported clinical adverse events observed in relation to any laboratory test values in this trial.
Acknowledgments
The other principal investigators in this multicenter study were the following: Robert W. Baker, M.D., Jackson, Miss.; Ileana Berman, M.D., Taunton, Mass.; Ronald Brenner, M.D., Far Rockaway, N.Y.; David Brown, M.D., Austin, Tex.; John S. Carman, M.D., Smyrna, Ga.; K.N. Roy Chengappa, M.D., Pittsburgh; Cal K. Cohn, M.D., Houston; John G. Csernansky, M.D., St. Louis; David G. Daniel, M.D., Falls Church, Va.; Michael DePriest, M.D., Salt Lake City; Louis Fabre, M.D., Houston; David Feifel, M.D., San Diego; James Ferguson, M.D., Salt Lake City; Michael Flaum, M.D., Iowa City, Iowa; John Gurkis, M.D., Pittsburgh; Evelyn Howanitz, M.D., East Orange, N.J.; Richard Josiassen, M.D., Norristown, Pa.; Philip Kanof, M.D., Tucson, Ariz.; Mary Ann Knesevich, M.D., Dallas; Alex Kopelowicz, M.D., Mission Hills, Calif.; John Lauriello, M.D., Albuquerque, N.Mex.; William Lawson, M.D., Indianapolis; Michael Lesem, M.D., Bellaire, Tex.; Douglas F. Levinson, M.D., Philadelphia; Jean-Pierre Lindenmayer, M.D., Wards Island, N.Y.; H.E. Logue, M.D., Birmingham, Ala.; Raymond C. Love, Pharm.D., Perry Point, Md.; Subramoniam Madhusoodanan, M.D., Far Rockaway, N.Y.; Theodore Manschreck, M.D., Fall River, Mass.; Joseph McEvoy, M.D., Butner, N.C.; Steven R. Marder, M.D., Los Angeles; Herbert Y. Meltzer, M.D., Nashville, Tenn.; Alan Mendelowitz, M.D., Glen Oaks, N.Y.; Hussam Mihtar, M.D., San Diego; Alexander Miller, M.D., San Antonio, Tex.; Raj Nakra, M.D., Chesterfield, Mo.; Jorg J. Pahl, M.D., Oklahoma City; Anand K. Pandurangi, M.D., Richmond, Va.; Michael Plopper, M.D., San Diego; John Prosser, M.D., Dallas; Narayana Reddy, M.D., Peoria, Ill.; P. Roy-Byrne, M.D., Seattle; Nina R. Schooler, Ph.D., Pittsburgh; S. Charles Schultz, M.D., Cleveland; Steven Strakowski, M.D., Cincinnati; Robert Stern, M.D., Bronx, N.Y.; Alan Swan, M.D., Houston; Steven Targum, M.D., Upland, Pa.; Dane Wingerson, M.D., Seattle; Donna A. Wirshing, M.D., Los Angeles.