Schizophrenia is associated with a two- to threefold greater risk of cardiovascular disease (CVD) compared with risk in the general population, contributing to a 10- to 20-year reduced life expectancy (
1,
2). Despite increasing awareness of this comorbidity over recent decades, the level of CVD risk does not appear to have decreased for individuals with schizophrenia (
3), and there has been little or no progress in decreasing the mortality gap (
2,
4,
5). The causes of the excess CVD risk remain unclear, but they are associated with lifestyle factors, antipsychotic use, inadequate somatic health care, and psychosocial challenges (
6–
8). In addition, a genetic susceptibility to CVD may play a role, as indicated by glucose disturbances in first-episode drug-naive patients with schizophrenia and increased CVD risk in first-degree relatives of individuals with schizophrenia (
9,
10). However, low body mass index (BMI) has been implicated as a risk factor for schizophrenia (
11). Moreover, there is considerable individual variation in other CVD risk factors, including hypertension, type 2 diabetes (T2D), and dyslipidemia (
3,
8,
12), which might suggest increased genetic liability to CVD in subgroups of patients with schizophrenia.
Schizophrenia is a complex polygenic disorder with an estimated heritability of 60%–80% (
13,
14). The largest genome-wide association study (GWAS) of schizophrenia to date identified 287 loci associated with the disorder (
15). GWASs have also detected several loci associated with coronary artery disease (CAD) (
16) and CVD risk factors, including BMI (
17), waist-to-hip ratio (WHR) (
18), T2D (
19), total cholesterol (TC) (
20), low-density lipoprotein (LDL) (
20), high-density lipoprotein (HDL) (
20), triglycerides (TG) (
20), systolic blood pressure (SBP) (
21), and diastolic blood pressure (DBP) (
21). Increasing evidence suggests genetic overlap between schizophrenia and CVD risk factors (
22–
26). For instance, genetic variants jointly associated with increased risk of T2D and schizophrenia have been identified (
23,
24). Studies have also revealed overlapping loci between schizophrenia and BMI, WHR, lipids, and SBP (
22,
25,
27). Interestingly, the majority of the shared loci between schizophrenia and BMI had opposite effect directions (
25), in line with a negative genetic correlation estimate (r
g=−0.11) (
25). However, there was an even distribution of concordant and opposite effect directions among the schizophrenia loci shared with lipids and SBP (
22). Similarly, we recently discovered shared variants between bipolar disorder and CVD risk factors that had a mixture of effect directions (
28). The pattern of mixed effects may indicate variation in genetic susceptibility to CVD across subgroups of patients (
28,
29).
Results
Genetic Overlap Between Schizophrenia and CVD Phenotypes
The univariate MiXeR results are presented in Table S100 in the
online supplement. The analyses revealed large differences in the number of variants accounting for 90% of SNP heritability across phenotypes: smoking initiation (N=11.1K), BMI (N=11.0K), and schizophrenia (N=9.6K) demonstrated the highest polygenicity, whereas SBP and DBP (N=4.4K and 3.9K), T2D (N=2.3K), lipids (N=0.8K to 1.8K), WHR (N=1.7K), and CAD (N=1.3K) showed lower polygenicity (
Figure 1; see also Table S100 in the
online supplement). SNP-based heritability, univariate Q-Q plots for observed versus predicted GWAS p values, and Q-Q plots partitioned by minor allele frequency and LD for each trait are presented in Figures S1–S2 and described in the Results section in the
online supplement. The univariate AIC and BIC values were all highly positive (see Table S100 in the
online supplement), indicating sufficient model fit.
The bivariate analyses indicated substantial genetic overlap between schizophrenia and some CVD phenotypes (
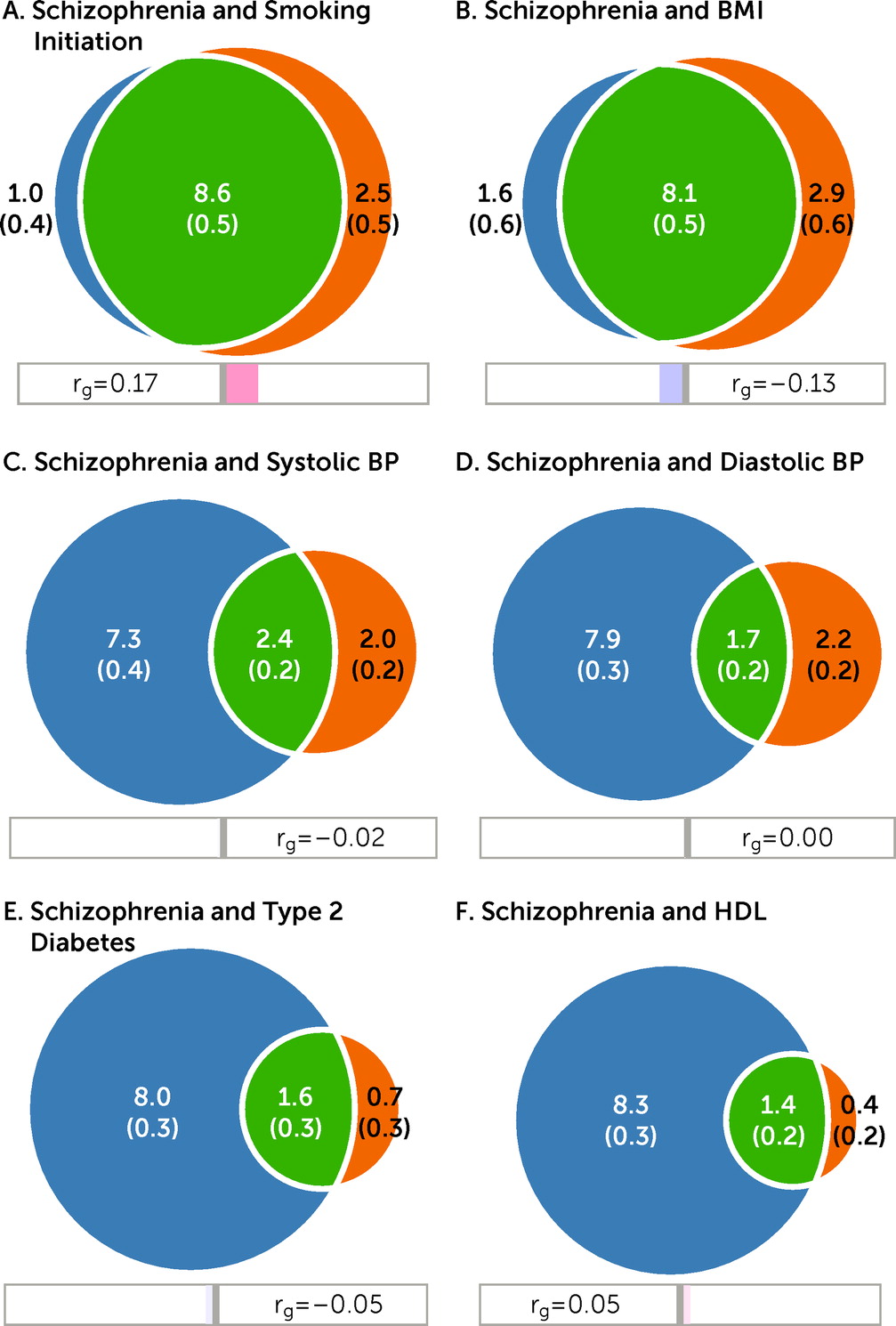
Figure 1). In particular, MiXeR estimated that of the 9.6K schizophrenia-influencing variants, 8.6K and 8.1K also influence smoking initiation and BMI, respectively (
Figure 1A–B). This corresponds to approximately 90% and 84% of schizophrenia-influencing variants overlapping with smoking initiation and BMI, respectively (
Table 1). Schizophrenia shared fewer variants with SBP (N=2.4K); DBP (N=1.7K); T2D (N=1.6K); lipids such as HDL (N=1.4K) (
Figure 1C–F), TG (N=1.2K), LDL (N=0.3K), and TC (N=0.3K); WHR (N=1.2K); and CAD (N=0.5K). These findings were in part because of lower polygenicity of these CVD phenotypes (see Figures S3–S10 in the
online supplement). Bivariate MiXeR was deemed to model the data adequately, as indicated by Q-Q plots, log-likelihood curves (see Figures S3–S10 in the
online supplement), and AIC values (see Table S101 in the
online supplement). The BIC values, which provide a stricter index of model fit than AIC values, were lower, but generally positive, indicating adequate model fit (see Table S101 in the
online supplement). MiXeR was not able to model cigarettes per day adequately, as shown in the log-likelihood curve in Figure S11 in the
online supplement. See the Results section in the
online supplement for further details.
The genetic correlations within the shared component (green areas in the Venn diagrams,
Figure 1) are presented in
Table 1. MiXeR estimated moderate positive genetic correlations within the shared component for schizophrenia and smoking initiation (r
gs=0.20), cigarettes per day (r
gs=0.36), LDL (r
gs=0.30), TC (r
gs=0.40), HDL (r
gs=0.15), CAD (r
gs=0.17), and WHR (r
gs=0.29), whereas negative genetic correlations were estimated within the shared component between schizophrenia and BMI (r
gs=−0.17) and T2D (r
gs=−0.13); the remaining correlations were close to zero (
Table 1). The genome-wide correlations (
Table 2) largely mirrored the correlations within the shared components (
Table 1), yet they were lower and nonsignificant for lipids, CAD, T2D, and WHR after correction for multiple testing.
Loci Shared Between Schizophrenia and CVD Phenotypes
After observing cross-trait enrichment in conditional Q-Q plots (see Figures S12–S13 in the online supplement), we applied a condFDR approach that identified several loci associated with schizophrenia conditional on CVD phenotypes (see Tables S1–S12 in the online supplement), including smoking initiation (N=362), SBP (N=325), DBP (N=317), TG (N=332), HDL (N=331), TC (N=272), LDL (N=279), cigarettes per day (N=307), WHR (N=303), and BMI (N=299). Next, we discovered multiple loci associated with CVD phenotypes conditional on schizophrenia (see Tables S13–S24 in the online supplement).
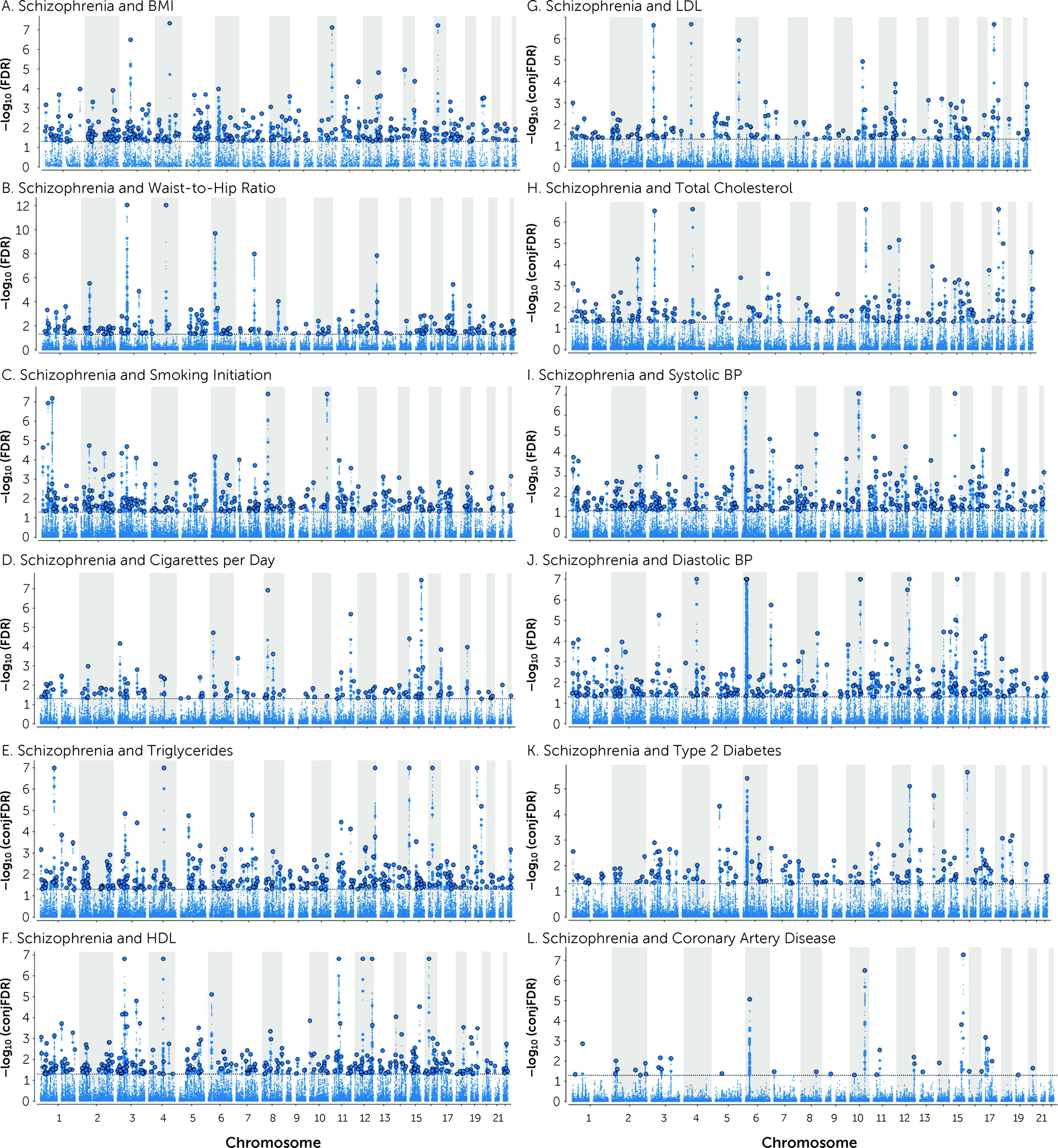
A total of 825 distinct loci were jointly associated with schizophrenia and CVD phenotypes at conjFDR<0.05 (
Figure 2 and
Table 2; see also Tables S25–S36 in the
online supplement). Schizophrenia shared 304 loci with BMI, 193 with WHR, 293 loci with smoking initiation, 129 loci with cigarettes per day, 307 with TG, 304 with HDL, 176 with TC, 158 with LDL, 294 with SBP, 259 with DBP, 147 with T2D, and 35 with CAD (
Figure 2 and
Table 2; see also Tables S25–S36 in the
online supplement). In addition, 357 loci were common for schizophrenia and more than one CVD phenotype (see Table S37 and Figure S14 in the
online supplement).
Of the loci associated with schizophrenia at condFDR<0.01, 104 are novel schizophrenia loci (see Table S38 in the online supplement). Of the shared loci at conjFDR<0.05, 348 are novel for schizophrenia (see Table S39 in the online supplement). This yielded a total of 366 novel schizophrenia loci with a condFDR<0.01 or conjFDR<0.05 (see Table S40 in the online supplement).
We determined the directionality of effects of lead SNPs within shared loci (
Table 2; see also Tables S25–S36 in the
online supplement): 35% were concordant for schizophrenia and BMI, 69% were concordant for schizophrenia and smoking initiation, and 63% were concordant for schizophrenia and cigarettes per day. Approximately half of the loci shared between schizophrenia and lipids, SBP, DBP, T2D, WHR, and CAD possessed concordant effect directions (
Table 2; see also Tables S25–S36 in the
online supplement). The effect directions broadly reflected the directions observed using genetic correlations (
Table 1 and
Table 2).
Validation: Consistency of Genetic Effects in Independent Samples
Validation analyses demonstrated a high degree of sign concordance (∼78%) between the independent discovery and replication data sets for the shared loci at conjFDR<0.05 (see Tables S41–S47 in the
online supplement). For exact binomial values, see the Results section in the
online supplement. The replication GWASs were considerably smaller than the discovery samples, which reduced the power to detect significant lead SNPs. Nevertheless, the consistency of associations in the replication data sets was comparable to that of other GWASs of complex traits (
39,
40), supporting the validity of these findings. Of the 304 lead SNPs in loci shared between schizophrenia and BMI, 73 and 74 had p values <0.05 in the replication samples for schizophrenia and BMI, respectively. Of the 294 lead SNPs in loci shared between schizophrenia and SBP, 58 and 91 had p values <0.05 in the replication samples, respectively. Of the 293 lead SNPs in loci shared between schizophrenia and smoking initiation, 44 and 40 had p values <0.05 in the replication samples, respectively. Of the 129 lead SNPs in loci shared between schizophrenia and cigarettes per day, 26 and 16 had p values <0.05 in the replication samples, respectively. Of the 307 lead SNPs in loci shared between schizophrenia and TG, 57 and 41 had p values <0.05 in the replication samples, respectively. Of the 304 lead SNPs in loci shared between schizophrenia and HDL, 50 and 41 had p values <0.05 in the replication samples, respectively. Of the 35 lead SNPs in loci shared between schizophrenia and CAD, nine and three had p values <0.05 in the replication samples, respectively. See the Results section and Tables S41–S47 in the
online supplement for more information.
Functional Annotation
Functional annotation of candidate SNPs shared between schizophrenia and CVD phenotypes demonstrated that they were mostly intronic and intergenic (see Tables S48–S59 in the online supplement). Next, we mapped candidate SNPs to genes using three gene-mapping strategies and discovered thousands of protein-coding genes (see Tables S60–S71 in the online supplement). Several SNPs were mapped to genes with chromatin interaction and eQTL associations in human brain tissue (e.g., fetal and adult cortex) and cells of the immune system (e.g., monocytes and CD4 T cells) (see the Results section and Tables S60–S71 in the online supplement).
The gene-set analysis of genes nearest to lead SNPs shared between schizophrenia and BMI implicated enrichment in several Gene Ontology (GO) terms, the most strongly associated terms being “regulation of transmembrane transport,” “regulation of synaptic plasticity,” and several neuronal gene sets (see Table S72 in the online supplement). There were no significant GO terms enriched among the genes nearest to lead SNPs shared with WHR, except a significant pathway termed “KEGG Alzheimer’s disease” (see Table S73 in the online supplement). Gene-set analysis of the genes nearest to the lead SNPs shared between schizophrenia and smoking initiation indicated GO terms with a predominance of gene sets related to neurodevelopment, including “central nervous system development” (see Table S74 in the online supplement). There were also overrepresented pathways among these genes, including “pathways affected in adenoid cystic carcinoma” (see Table S74 in the online supplement). The enriched gene sets associated with schizophrenia and lipids, SBP, and DBP involved neuronal, synaptic, and immunological gene sets (see Tables S75–S79 in the online supplement). In addition, there were significantly enriched gene sets associated with DNA-binding and cellular processes (see Tables S75–S79 in the online supplement). Pathway analysis of the genes mapped to schizophrenia and TG, SBP, and DBP indicated “MAPK signaling pathway,” “brain-derived neurotrophic factor signaling pathway,” and “energy metabolism” (see Tables S75, S78, and S79 in the online supplement). Moreover, the enrichment analyses including genes in MHC and 8p23.1 yielded results similar to those excluding these genomic regions (see Tables S72–S79 in the online supplement). This indicates that these genomic regions do not have a substantial impact on the results.
We also performed gene-set and pathway analyses with genes mapped to all candidate SNPs. The results agreed broadly with the results based on genes nearest to lead SNPs, yet were less specific and included several intra- and intercellular gene sets (see the Results section and Tables S80–S98 in the online supplement).
Genetic Overlap Between CVD Phenotypes
The genetic correlations between the CVD phenotypes ranged from low to high, with the strongest correlations between TC and LDL (rg=0.946, p=3.35×10−12) and between SBP and DBP (rg=0.808, p<1.00×10−20) and the weakest between lipids and blood pressure (rg=−0.01 to −0.12, p=0.62 to 1.54×10−9) (see Figure S15 and Table S99 in the online supplement). MiXeR analyses demonstrated different levels of genetic overlap, with the most overlap between SBP and TG, BMI and CAD, BMI and TG, and BMI and T2D (see Figure S16 in the online supplement). Adequate MiXeR model fit, however, was restricted to BMI and smoking initiation, CAD and TG, SBP and TG, CAD and SBP, SBP and smoking initiation, SBP and T2D, and BMI and SBP (see Figure S16 and Table S102 in the online supplement). The other bivariate estimates were uncertain, as indicated by no clear minimum on the log-likelihood curves, Q-Q plots (the observed Q-Q plots did not closely follow the model predictions), and negative AIC and BIC values (see Figure S16 and Table S102 in the online supplement).
Genomic Structural Equation Modeling Results
We further explored the genetic relationship between schizophrenia and CAD controlling for the effect of risk factors (smoking initiation, BMI, HDL, SBP, and T2D). The multiple regression analyses demonstrated no statistically significant impact of these CVD risk factors, except for a minor effect of smoking initiation on the relationship between schizophrenia and CAD, resulting in a slightly modified genetic correlation between schizophrenia and CAD (rg=−0.055, p=0.041) (see Figure S17 in the online supplement). Mediation analyses, however, found statistically significant effects for all CVD risk factors except SBP (see Figure S18 in the online supplement), although the indirect effect between schizophrenia and CAD remained close to zero (rg=−0.035 to 0.036), indicating minor effects. For further details, see the Results section and Figures S17–S18 in the online supplement.
Discussion
In this study, we found that a considerable proportion of genetic variants underlying schizophrenia also influence CVD phenotypes, particularly the risk factors smoking initiation and BMI, using MiXeR. Next, we detected more than 800 distinct loci jointly associated with schizophrenia and CVD risk factors and CAD at conjFDR<0.05. Most of the loci shared between schizophrenia and smoking initiation and cigarettes per day possessed concordant effect directions, in line with positive genetic correlations. The overlapping loci with BMI had mainly opposite effect directions, consistent with negative genetic correlations. There was a pattern of mixed effect directions among loci jointly associated with schizophrenia and the other CVD phenotypes, including lipids, SBP, DBP, T2D, WHR, and CAD. The high degree of sign concordance between the discovery and replication samples supports the validity of the findings. Furthermore, functional analyses of the shared loci implicated genes associated with neurodevelopment and the immune system. These results shed light on putative biological functions and pathways associated with the comorbidity between CVD and schizophrenia that warrant further investigation and experimental validation.
The MiXeR analyses indicated differences in the level of genetic overlap across various CVD risk factors and CAD. In particular, MiXeR estimated that the majority of the SNPs influencing schizophrenia also affect smoking initiation (∼90%) and BMI (∼84%), whereas less genetic overlap was observed with the other CVD risk factors. Smoking and BMI seem to be more polygenic than the other CVD phenotypes. The causes of the differences in polygenicity are unclear but may be related to smoking and BMI being more influenced by behavior that is regulated by the brain than the other CVD phenotypes. Other analyses have also revealed that behavioral phenotypes have higher polygenicity than somatic diseases and traits (
57). Lower polygenicity of the other CVD phenotypes compared with BMI and smoking initiation limits the genetic overlap with schizophrenia.
Our conjFDR analyses detected several genetic loci jointly associated with schizophrenia and CVD risk factors, especially BMI, smoking initiation, blood pressure, and TG. Larger GWAS samples of these CVD phenotypes compared with the GWASs of T2D and CAD are likely to contribute to the greater number of identified loci. A total of 366 of the identified loci are novel to schizophrenia. The results illustrate the increased SNP discovery by leveraging pleiotropy with CVD by using the condFDR and conjFDR approaches (
22,
32). The allelic effect directions of the shared loci identified by conjFDR were mainly in line with the genome-wide correlation and genetic correlations within the shared components obtained from MiXeR. Note, however, that the identified loci at conjFDR<0.05 represent only a small fraction of the genetic architecture, and the concordance in allelic effects among these shared loci may not necessarily align with the measures of genetic correlation at the genome-wide level or within the shared components.
Furthermore, the majority of the shared loci have opposite allelic effect directions in schizophrenia and BMI. The results indicate that people with schizophrenia are genetically predisposed to lower BMI, at the group level. This is in line with previous genetic findings (
25) and clinical evidence of low BMI being a risk factor for schizophrenia (
11) and being more prevalent among individuals with schizophrenia than in the general population (
58,
59). However, obesity is also more common in individuals with schizophrenia compared with those in the general population (
3,
58). The present findings indicate that factors other than common genetic variants play an important role in weight gain in schizophrenia (
25). In particular, adverse effects of antipsychotics and unhealthy lifestyle related to negative symptoms, depression, and socioeconomic challenges are likely to be major causes (
6–
8,
60,
61). However, genetic factors appear to play an important role in antipsychotic-induced weight gain as well, with a heritability estimate of 60%–80% (
62). Taken together, the evidence indicates interactions between genetic liability to antipsychotic-induced weight gain, lifestyle factors, and psychosocial challenges as underlying mechanisms of weight gain.
Despite the negative genetic correlation between BMI and schizophrenia, a trend toward a positive association was observed for WHR (r
g=0.039). Although this may seem inconsistent with the inverse schizophrenia-BMI association, WHR correlates weakly with BMI clinically (
63), and WHR adjusted for BMI demonstrated no significant genetic correlation with BMI in our study (see Table S99 in the
online supplement). Moreover, the genetic correlation between schizophrenia and WHR was not significant after correction for multiple testing, in line with previous studies (
27,
64), although one study found a statistically significant, yet low, negative genetic correlation between schizophrenia and waist circumference (r
g=−0.07) (
65). Furthermore, the results indicate that the higher WHR seen in antipsychotic-naive patients with schizophrenia (
66) is not due mainly to genetic factors, but rather to lifestyle factors.
We discovered extensive genetic overlap between schizophrenia and smoking behavior. The effect directions of the shared SNPs are in line with the moderate positive genetic correlation estimated here and in previous studies (
30,
31). The results indicate an increased genetic propensity for smoking associated with schizophrenia. The addictive properties of nicotine may have a larger influence in people with schizophrenia, in line with evidence of greater nicotine dependence among individuals with schizophrenia than in the general population (
67). In particular, patients with schizophrenia experience greater reinforcing effects of nicotine and more severe withdrawal symptoms during abstinence (
67,
68). The higher nicotine dependence in schizophrenia may be partly genetically driven, as indicated by a positive genetic correlation (
69). Moreover, studies have linked both nicotine dependence and schizophrenia to variants in the nicotinic acetylcholine receptor (nAChR) gene cluster (
CHRNA3-
CHRNA5-CHRNB4) (
15,
69). Here, we corroborated associations of variants in the nAChR gene cluster with schizophrenia and cigarettes per day (see Table S51 in the
online supplement). In addition, smoking may represent a form of self-medication. Nicotine activates nAChRs, which stimulates release of dopamine, serotonin, and glutamate (
68). Thus, tobacco smoking in people with schizophrenia may involve, to some extent, an attempt to compensate for genetically determined dysfunction of nAChRs, implicating monoaminergic and glutamatergic signaling (
14,
68), although this requires further investigation.
We identified several shared loci with mixed effect directions in schizophrenia and lipids, SBP, DBP, WHR, T2D, and CAD. The results validate earlier findings of bidirectional effects among overlapping loci between schizophrenia and CVD risk factors (
22,
26,
27) and between bipolar disorder and CVD risk factors (
28). Moreover, the present study extends previous results (
22) by providing a more detailed characterization of the shared genetic architecture and specific overlapping loci. Nevertheless, the common SNPs identified here and previously (
22,
26–
28) have small effects, explaining a small portion of the overall disease risk. The remaining variance is likely to be explained by multiple undetected SNPs, rare variants, interactions between genes, and interactions between genes and environmental factors, especially antipsychotics and unhealthy lifestyle (
6,
60).
Although the average level of CVD risk is higher in schizophrenia compared with the general population, there is considerable individual variation in CVD risk (
3,
8,
12). Our findings of shared loci with mixed effect directions may reflect variation in genetic susceptibility to CVD across subgroups of schizophrenia. Thus, there may be subsets of patients with a higher genetic liability to CVD, as indicated by previous studies (
29,
70). This possibility can help explain reports of cardiometabolic disturbances in drug-naive patients with schizophrenia at illness onset and among their relatives compared with healthy control participants (
9,
10,
71). Moreover, a recent study provided evidence of genetic contribution to T2D in schizophrenia (
24), although findings have been inconsistent (
15,
72). Patients with more negative symptoms may represent a subgroup who are at greater risk for CVD, consistent with findings suggesting that patients with more severe and enduring negative symptoms have a higher CVD risk, whereas positive psychotic symptoms seem to be less associated with CVD (
73,
74). Larger GWAS samples with phenotypic refinement are required to identify potential subgroups of schizophrenia patients with differential liability to CVD.
Both overweight and smoking are risk factors for CAD, in line with their positive genetic correlation with CAD shown here. We investigated whether BMI and smoking initiation, which have a negative and positive genetic correlation with schizophrenia, respectively, influence the genetic relationship between schizophrenia and CAD. We hypothesized that controlling for these CVD risk factors would modify the association between schizophrenia and CAD. The results from the genomic structural equation modeling suggested minimal effect on the relationship between schizophrenia and CAD, indicating that there are other factors explaining the nonsignificant correlation between them. Furthermore, we did not find support for a systematic change in the direction of the association between schizophrenia and CAD by controlling for the other CVD risk factors. These findings support a mixture of effect directions across the different CVD risk factors. Thus, it does not appear as though controlling for the effect of a CVD factor (e.g., smoking) affects a subgroup of variants that are shared with schizophrenia and CAD. However, the statistical power for finding such a pattern may have been inadequate.
Functional analyses of the shared loci implicated genes associated with neurodevelopment, synaptic function, immune system, intra- and intercellular processes, and metabolic mechanisms. The results are in line with the neurodevelopmental hypothesis and with the immune system being implicated in the pathogenesis of schizophrenia (
14). In addition, brain function regulates behavior, which plays a key role in lifestyle habits influencing CVD. The immune system also plays a crucial role in the development of CVD (
75). The findings from the functional analyses align with recent studies of genetic overlap between schizophrenia, bipolar disorder, and CVD risk factors (
25,
28,
64). However, the results should be considered with caution given the limitations of functional annotation methods (
37) and the complex mechanisms underlying schizophrenia and comorbid CVD. The methods used in the present study are limited by uncertainties in translating genetic loci to causal variants, which restricts the biological interpretation of the shared genetic variants (
14). Some additional methodological limitations should be noted. The model underlying MiXeR is sensitive to LD structure estimates. Discrepancies between the LD structure of the samples used for the GWAS and that of the reference panel may have biased the model’s estimates. Additionally, implementation of the MiXeR model assumes similar LD structure in the analyzed GWAS, which hinders the analysis of GWASs based on genetically dissimilar populations, including transancestry analyses. Thus, we are working on implementation that supports transancestry analyses, permitting transferability of results across ethnicities.
In summary, we revealed polygenic overlap between schizophrenia and CVD risk factors, particularly BMI and smoking. The results indicate an inherent propensity to smoking in individuals with schizophrenia. In contrast, several schizophrenia risk loci appear to be protective against obesity. The findings highlight the importance of environmental factors in the development of obesity and other CVD comorbidities. In addition, the mixed effect directions of the shared loci between schizophrenia and lipids, blood pressure, T2D, and CAD may suggest variation in genetic vulnerability to CVD across schizophrenia subgroups, which may underlie some of the observed heterogeneity in CVD comorbidity. Studies with larger GWASs will uncover more of the genetic architecture of schizophrenia and may reveal differences in genetic liability to CVD across subsets of patients. Such findings could provide clinically useful discoveries that pave the way for risk stratification and more tailored interventions. Further work is needed to identify the causal genetic variants and determine their functional properties. This research is likely to provide insights into the mechanisms underlying the comorbidity and might facilitate the development of antipsychotics with lower metabolic side effects. Such progress will enable more effective prevention of comorbid CVD, thereby helping to mitigate a major clinical and health care problem.