In 1980 Coulter and Allen (1) reported the first case of hyperammonemia, to our knowledge, with otherwise normal hepatic function tests in a child with epilepsy treated with valproic acid. Since that time, there have been several additional case reports and studies within the neurology literature that have established hyperammonemia with otherwise normal hepatic function as a potential side effect of valproic acid, especially in children and adolescents (2) . Settle (3) published the first case of hyperammonemia, to our knowledge, in a psychiatric setting in 1995. Several additional cases have followed it. Unfortunately, to date, to our knowledge, there has only been one prospective study published within the psychiatric literature (4) .
We present here a patient’s history that illustrates how the symptoms of a psychiatric disorder might make the diagnosis of hyperammonemic encephalopathy difficult.
Case Presentation
“Ms. A” was a 45-year-old woman with schizoaffective disorder, bipolar type, beginning in her teens. Her psychiatric history included multiple prior hospitalizations for psychosis. Over the years, she had been stabilized with various medications, including valproic acid. During her most recent hospitalization, about 1 year earlier, she had been stabilized with carbamazepine and risperidone. She had done well with this combination until her mother passed away 6 months before her current admission. Her mother had also been her primary caregiver and was the best source of information about her psychiatric history. Following her death, Ms. A’s brother assumed some responsibilities for her, including informed consent to give medications when she lacked the capacity.
Her medical history was significant for chronic obstructive pulmonary disease and poorly controlled high blood pressure with associated dilated cardiomyopathy. Although her history included congestive heart failure during an episode of pneumonia, an echocardiogram during this admission revealed an ejection fraction of 40% and well-compensated heart failure. She had no history of neurological problems, specifically seizures or unexplained changes in level of consciousness. Her medications included a fluticasone and salmeterol combination inhaler b.i.d., an albuterol and ipratropium combination inhaler four times a day, and furosemide, 20 mg/day, all of which appeared to be controlling her pulmonary and cardiac problems.
Following the death of her mother, Ms. A required involuntary admission to the psychiatric unit of a local hospital. During her stay there, her antipsychotic regimen was increased to include three medications: risperidone, 3 mg b.i.d.; fluphenazine, 15 mg b.i.d.; and quetiapine, 400 mg at bedtime. However, according to transfer records, Ms. A never reported a lessening in her auditory hallucinations and was never able to perform her own activities of daily living independent of the nursing staff. She was transferred to our facility for refractory psychosis after 1 month of unsuccessful inpatient treatment.
Upon transfer, Ms. A’s weight was 61.8 kg. She was disheveled and showed little evidence of self-care. She continued to report auditory hallucinations, frequently a running conversation that was giving negative descriptions of her and her behavior. She also exhibited a loosening of associations and a labile affect with outbursts of either tears or laughter that were incongruent with her reported mood. However, she was fully oriented and able to describe her treatment options and was deemed to still have capacity to give informed consent for medications. She requested to restart valproic acid and to take risperidone as her only antipsychotic, a combination that had worked well for her during a prior hospitalization in our facility. Her antipsychotic regimen was reduced to risperidone, 3 mg b.i.d., to target her continued disorganized thoughts and auditory hallucinations. Valproic acid, 1000 mg (16.2 mg/kg) every evening, was added for mood stabilization and was increased 3 days later to 1500 mg (24.3 mg/kg) every evening.
For the first six days of hospitalization at our facility, Ms. A was cooperative and improved in her level of self-care. However, by day six, she became agitated and required emergency antipsychotic medication to maintain her own safety. She also appeared confused at times and was no longer able to participate in the ward milieu. Her brother was then appointed as her legally authorized representative for informed consent for medications. On the 10th day of hospitalization, she had her first episode of severely decreased level of consciousness. She was found by staff lying on her bed. She was unresponsive but her eyes were blinking slowly, foam was in her mouth, and she had bladder incontinence. There was no evidence of a tongue bite. She was transported to the nearest emergency room to be evaluated for a possible seizure. An evaluation in the emergency room found a supratherapeutic valproic acid level of 141 mg/ml, but otherwise Ms. A was awake, responsive, laughing at times, and appeared psychotic to the emergency room staff. She was diagnosed with a pseudoseizure and returned to the psychiatric ward. Her valproic acid dose was reduced to 1000 mg/day.
About 12 hours after returning from the emergency room, her roommate witnessed another possible generalized seizure, described as “shaking all over.” Ward staff found Ms. A lying in bed with her eyes open but unresponsive and with loud labored breathing. She again had foam in her mouth and was difficult to arouse. She appeared confused and lethargic, and she vomited when staff raised her to a seated position. She was again transported to the emergency room, and this time she was admitted to an internal medicine service for evaluation of repeated episodes of loss of consciousness. She underwent a brain computerized tomography scan without contrast, which was normal. Her laboratory studies, including a CBC, liver function tests, levels of serum glucose, protein, albumin, cardiac enzymes, and electrolytes, including sodium, were normal. A cardiology consult, which included an ECG, found no evidence of current congestive heart failure and no cardiac involvement in her altered level of consciousness. Furosemide was subsequently discontinued. A neurology consultant diagnosed Ms. A with “two episodes of unresponsiveness of unclear etiology” and stated, furthermore, that “her history of schizophrenia may be related to the events.” A brain magnetic resonance imaging (MRI) and echocardiogram were recommended. However, Ms. A refused the MRI, and she pulled the EEG electrodes off of her scalp. After two days in the internal medicine service, Ms. A had no further episodes of altered consciousness and was transferred back to the psychiatric ward.
Over the next seven days, Ms. A’s thoughts remained disorganized, and she was unable to participate in the milieu. She exhibited fluctuating confusion and disorientation, and she also began to complain of “seeing devils and angels on my shoulder” in addition to her continued auditory hallucinations. Her trough valproic acid level 1 week after the previous supratherapeutic check was therapeutic at 114 mg/ml.
Three weeks after starting valproic acid and 2 weeks after her first documented episode of altered consciousness, a venous serum ammonia level was checked and found to be 445 mg/dl (reference range=19–87). Valproic acid was immediately discontinued, and Ms. A was given a one-time dose of lactulose, 60 mg. Twenty-four hours later, her ammonia level was 158 mg/dl, and it normalized to 49 mg/dl 4 days later. It was normal throughout the rest of her remaining 2 months in our hospital. Additionally, her fluctuating level of consciousness resolved with the reduction in her hyperammonemia and did not return during her hospitalization. Ms. A no longer complained of visual hallucinations or disorientation but remained disorganized and exhibited mood lability. She was then treated with another mood-stabilizing medication, which she tolerated better. She continued to take risperidone without further hyperammonemia. She was not rechallenged with valproic acid during her hospitalization. As she recovered, she was able to provide some additional history. She reported no prior history of seizures, and her brother confirmed this statement. She did tell the treatment team that she had recently become a strict vegetarian, but her brother could not verify this history. Additionally, her brother reported no family history of known inborn errors of metabolism, such as boys dying in infancy suspected for X-linked ornithine transcarbamylase deficiency, and no family members with a history of seizures or unexplained altered levels of consciousness.
A Literature Review of Hyperammonemia Due to Valproic Acid
MEDLINE was searched from 1966 to March 2007. The MeSH headings were “valproic acid,” “encephalopathy,” and “hyperammonemia.” Bibliographies of identified articles were further reviewed. There was extensive literature on this phenomenon within pediatric neurology, but our review focused on studies involving valproic acid in a psychiatric setting. There were no limits placed on the age of the patient. Direct contact was attempted with the authors of the pediatric studies for any additional unpublished material. There were no randomized or blinded studies found within the psychiatric literature.
The Only Cohort Study on Hyperammonemia
To our knowledge, only one prospective study has been conducted on hyperammonemia due to valproic acid in a psychiatric setting. In it, Raja and Azzoni (4) assessed consecutive adult patients who were admitted to a psychiatric unit and treated with a mood stabilizer for the presence of hyperammonemia. They found that 51.2% of the patients receiving valproic acid (N=123) had asymptomatic hyperammonemia (level >97 mg/dl) as did 21.7% in the control arm (N=23), which is a greater prevalence than that found in a pediatric neurology study they referenced (5) . Raja and Azzoni (4) also found a positive correlation between valproic acid serum concentration and ammonia level. The mean ammonia level was higher in the valproic acid group than the control group, and liver function remained normal in the patients with elevated ammonia. Instead of discontinuing valproic acid use upon the discovery of hyperammonemia, they only lowered the dose. This reduction in dose always resulted in a reduction in ammonia level for their patients. However, the authors gave no specific data for these comments. They surmised that complete withdrawal of the medication is warranted in only the most severe cases of side effects. Raja and Azzoni also note that mental status changes due to valproic acid are difficult to distinguish from worsening psychosis or mania or even from a therapeutic response. They conclude with recommendations to monitor both liver function and serum ammonia in patients taking valproic acid to assist in the early detection of adverse effects (4) .
Raja and Azzoni attributed the higher prevalence of hyperammonemia than that found in pediatric neurology populations to either the age difference itself, the presence of other psychotropic medications, or alcohol intake in their population. However, the study the authors cite by Bohles et al. (5) was designed to look at carnitine supplementation instead of screening for the prevalence of hyperammonemia. Raja and Azzoni (4) cite several limitations to their study, including the nonrandom assignment of patients, the lack of blinded assessment, the lack of standardized assessment, a small control group, each variable being measured multiple times, and no screening for heavy alcohol use. Some additional limitations included a lack of assessment for risk factors for hyperammonemia, no stratification based upon length of use of valproic acid, no comment on possible symptoms due to hyperammonemia, and blood levels being drawn only one time close to admission. Thus, although this study gives an initial indication that some psychiatric patients develop hyperammonemia, the authors were unable to comment on the importance of this finding, the possible risk factors that might explain its presence, or the impact of it on patients.
Case Reports of Encephalopathy
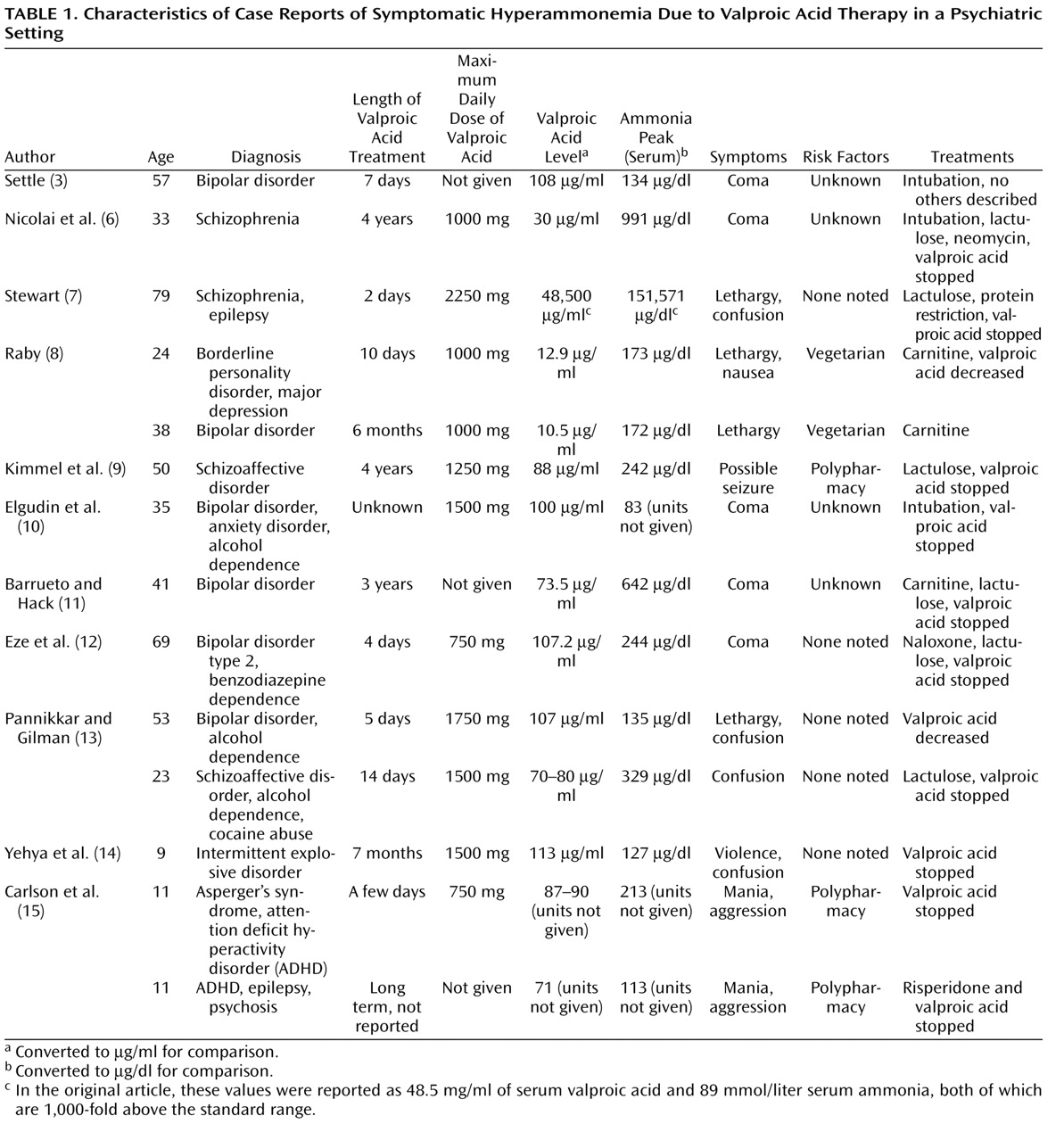
The 11 case reports of symptomatic hyperammonemia in a psychiatric setting are listed in Table 1 . They describe 14 patients with various psychiatric disorders, demonstrating both Food and Drug Administration approved and off-label uses of valproic acid. In one case, valproic acid was prescribed as a prophylaxis against seizures in clozapine therapy (6) . A patient in one of the reports had epilepsy, which was stable and being treated with phenytoin when valproic acid treatment began (7) . In reporting plasma valproic acid and ammonia levels in milligrams and millimoles instead of micrograms and micromoles, respectively, the case report by Stewart placed data for both one thousand times outside of the normal range (7) . None of the studies specified the form of valproic acid prescribed.
The authors of three of the cases identified potential risk factors from their patients’ histories. In one of these studies, Raby (8) noted that both patients had been vegetarians before their treatments, which the author linked to decreased carnitine levels. Kimmel and colleagues (9) hypothesized that polypharmacy played a role in their patient’s hyperammonemia, specifically an interaction between lorazepam and valproic acid. Many of the writers speculated, however, that perhaps their patients had an undiagnosed underlying genetic disorder placing them at risk for hyperammonemia, such as a urea cycle disorder or a carnitine disorder. Only one of the patients in the case reports was tested for evidence of a urea cycle disorder, and the tests were negative (6) . In four of the other cases, most of the patients’ medical or psychiatric histories were not known at the time of treatment. Their risk factors were listed as “Unknown” in Table 1(3, 6, 10, 11) . These were also four of the five patients who developed a coma.
Reported symptoms due to hyperammonemia in most of the cases involved confusion, lethargy, and other symptoms of encephalopathy. No authors found any evidence of hepatic injury or failure in their patients. Five of the 14 patients developed a coma (3, 6, 10 – 12 ). Four of these five comatose patients required intubation and admission to a medical intensive care unit. Three were first seen in the emergency room for severe mental status changes (6, 10, 11) . The two remaining patients that developed a coma were being treated on an inpatient psychiatric unit (3, 12) . However, the physicians there were not their primary psychiatrists, and the history of at least one of the two was “obscure” (3) .
All but one of the 12 patients recovered from encephalopathy less than 2 weeks after initiating treatment for it, usually within a few days. The two patients who were maintained with valproic acid and were treated with carnitine supplementation improved after 10–14 days (8) . The one patient in which encephalopathy did not resolve within 2 weeks remained in an intensive care unit for 26 days, but this case was also complicated by simultaneous diabetic ketoacidosis (6) . This patient was also the only one given an antibiotic—neomycin—specifically as a treatment for hyperammonemia to reduce bacterial production of ammonia in the gastrointestinal system.
Symptoms of Valproic Acid-Induced Hyperammonemia
The prevalence of symptomatic hyperammonemia leading to encephalopathy from valproic acid is unknown but thought to be quite rare in adults. Case reports on adults are almost always in specific at-risk populations or circumstances. The symptoms accepted within the neurological literature for encephalopathy induced by valproic acid include acute onset of impaired consciousness and lethargy, focal neurological signs or symptoms, and increased seizure frequency (16) . Other reported symptoms include asterixis, vomiting, perseveration, aggression, ataxia, and eventually coma and death (1, 17) . EEG findings, performed in a few patients with preexisting epilepsy, included a pronounced general slowing, an increase in epileptiform discharges, and possibly the presence of triphasic waves (16, 18) .
There remains controversy as to whether these symptoms have any relationship to daily dose or plasma concentration of valproic acid. Multiple studies, a few randomized but most of them cohort studies, have searched for a correlation between dose of valproic acid or plasma valproic acid concentration and serum ammonia levels. The results are mixed. Hyperammonemia occurs at both therapeutic and supratherapeutic concentrations of valproic acid, implying that other factors often influence the development of symptomatic hyperammonemia. There also appears to be no reassurance in prior safe treatment with valproic acid. According to Verrotti et al. (16), symptoms of encephalopathy may develop in someone who has previously taken it without complication.
Our patient exhibited many symptoms consistent with encephalopathy, including an acute change in her level of consciousness, confusion, nausea, and vomiting. After beginning valproic acid, several cues arose that encephalopathy might have been superimposed over her psychosis. Her confusion and disorientation exhibited a fluctuating course and were more consistent with delirium than disorganized psychosis. Her hallucinations changed, becoming visual in addition to auditory. Additionally, our patient’s symptoms of frothing at the mouth, incontinence, and a decreased level of consciousness on two occasions were consistent with possible seizures. Unfortunately, no EEG during this time period was obtained. There is only one other case report in the English literature, to our knowledge, of a possible seizure due to hyperammonemia from valproic acid in a patient without an underlying seizure disorder. It is also from a psychiatric setting, and no EEG was obtained in that case either (9) .
Proposed Mechanisms of Action
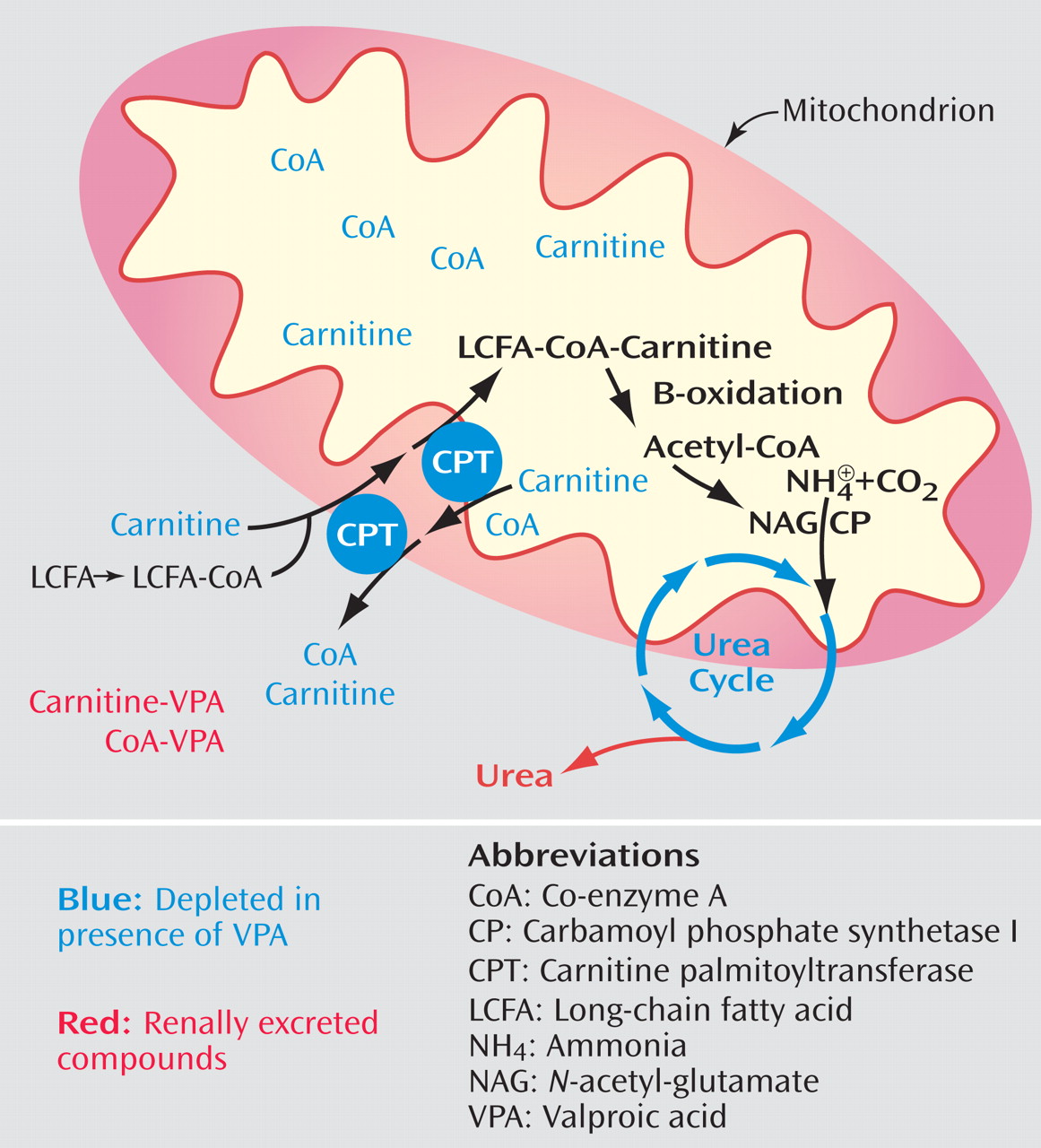
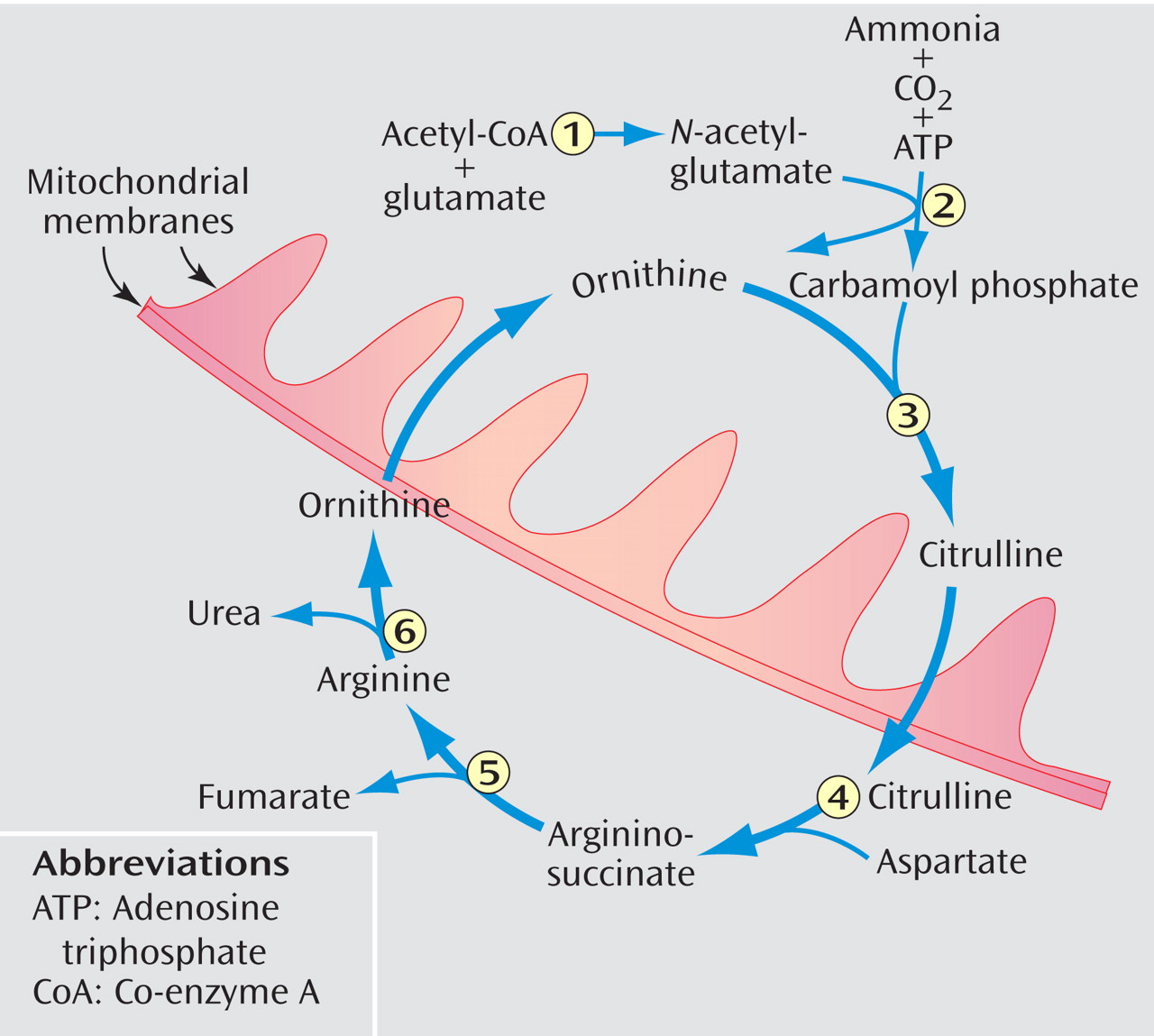
Various etiologies for hyperammonemia induced by valproic acid have been explored, but it is still not completely understood. Animal studies have found both a renal and hepatic role, with the latter appearing to be the dominant source (16, 19) . In the kidney, valproic acid and its derivatives are thought to affect renal uptake of glutamine, which increases ammonia production (19) . The potential mechanisms within the liver that lead to increased serum ammonia are depicted in Figure 1(16, 20) . These actions in the liver ultimately affect the urea cycle, which is shown in Figure 2 . An association between encephalopathy induced by valproic acid and genetic impairment in the urea cycle was described in the pediatric neurology literature, the age when most of these genetic disorders are first recognized (21) . In 2002 the Food and Drug Administration added a contraindication into the package insert of valproic acid against the use of it in patients with known urea cycle disorders and also a warning of the possibility of hyperammonemia in the presence of normal liver function tests (22) .
Figure 1. Potential Liver Mechanisms That Lead to Increased Serum Ammonia Levels a
a In hepatic mitochondria, valproic acid is believed to cause an accumulation of ammonia by reducing free carnitine and co-enzyme A, which bond to valproic acid and are then either renally excreted or sequestered in the mitochondrial matrix. Loss of carnitine prevents importation of long-chain fatty acids into the mitochondrial matrix for metabolism. Loss of co-enzyme A prevents the beta-oxidation of fatty acids into acetyl-co-enzyme A, which is a substrate of N -acetyl-glutamate, the required activator of the initial enzyme in the urea cycle.
Figure 2. Valproic Acid and the Urea Cycle a
aN -acetyl-glutamate, which valproic acid is thought to deplete, is a required activator of carbamoyl phosphate synthetase I. Enzymes of the cycle are the following: 1) N -acetyl-glutamate synthetase, 2) carbamoyl phosphate synthetase I, 3) ornithine transcarbamylase, 4) argininosuccinate synthetase, 5) argininosuccinate lyase, and 6) arginase. Both ornithine and citrulline are transported across the mitochondrial membranes.
Brusilow (23), in a narrative review on hyperammonemia, summarized how ammonia is thought to cause encephalopathy. Within the CNS, increased ammonia leads to higher production and accumulation of glutamine within astrocytes. This increased intracellular glutamine leads to cerebral edema and astrocyte dysfunction. The mechanisms by which the brain is thought to compensate for astrocyte swelling in chronic hyperammonemia include decreased osmolarity and thus edema by down-regulation of myo -inositol, increased brain tissue compliance, and mild to moderate brain atrophy (23) .
Asymptomatic Hyperammonemia
Valproic acid has been associated with chronic asymptomatic hyperammonemia. Several early studies demonstrated the presence of asymptomatic hyperammonemia in epileptic patients treated with valproic acid (24) . Several cohort studies in the neurological literature examined the prevalence of asymptomatic hyperammonemia in patients taking valproic acid. One early study by Murphy and Marquardt (25) found a 53% prevalence among pediatric patients taking valproic acid for various seizure disorders versus a prevalence of zero among their control subjects. A later study by Altunbasak et al. (2) found much lower rates: 5.6% of those taking valproic acid alone and 16.2% of those receiving polypharmacy among a pediatric population. Various authors attribute this prevalence range in the neurological literature to differences in the age of patients, polypharmacy, normal diurnal variation in serum ammonia, and the timing of blood drawn in relation to meals. Although the majority of studies have been done in pediatric populations, one smaller study on adults with mental retardation found asymptomatic hyperammonemia in 11 of 19 patients (58%) (26) .
Risk Factors
In the neurological literature, prospective studies and case reports identified several possible risk factors for hyperammonemia. The focus was placed on urea cycle disorders, infancy (immature hepatic function), and carnitine deficiency due to either genetic abnormalities or dietary restrictions (21) . Other researchers have focused on the effects of nutritional intake, polypharmacy, and complicating medical conditions. Studies found increased serum ammonia while subjects were taking valproic acid with either low caloric intake, higher nitrogen load (27), or possibly increased carbohydrate intake (28) . Within the neurological literature, instead of general medical conditions, the focus was on the coexistence of multiple neurological problems or the need for parenteral nutrition (29) . Any medical conditions that lead to a poor nutritional status or a catabolic state were also thought to put patients at risk for hyperammonemia. This risk factor was thought to act through carnitine deficiency. Poor nutrition or an idiosyncratic diet is common among people with severe mental illness, and our patient reported impulsively becoming a vegetarian as her mental health deteriorated in the months just before her hospitalization.
A well-established risk factor for hyperammonemia in the neurological literature is the combination of valproic acid with other antiepileptic medications, particularly phenobarbital and phenytoin. Initial studies in the early 1980s identified a link between administration of phenobarbital and valproic acid (30) and the administration of valproic acid with phenobarbital and/or phenytoin (31) . The mechanisms of action are not well understood but are thought to be related to either an increase in the production of toxic valproic acid metabolites through beta-oxidation or a synergistic effect on the urea cycle (31) . Case reports have also raised the possibility of an interaction between the effects of valproic acid and topiramate that leads to hyperammonemia, particularly topiramate’s inhibition of glutamine synthetase in the CNS (32) .
In Table 1, the most recently published case report implicates an interaction between valproic acid and risperidone that raises plasma levels of valproic acid, possibly through competition for protein binding sites in the blood, and thus increases the risk of hyperammonemia (15) . Although the U.S. prescribing information for divalproex sodium does not list this interaction (33), it is described in the package insert for risperidone. There, a study shows that this combination had no effect upon average plasma concentration or exposure (area under the curve) of valproic acid but did raise the peak concentration (C max ) of valproic acid by 20% (34) . However, the effect of this exposure to a higher peak plasma level of valproic acid has not been fully elucidated, and some studies, in fact, dispute the clinical significance of this interaction (35) . Our patient was also taking this combination and had done so while previously at our hospital without complication. At a weight of 61.8 kg, our patient received an initial dose of 16.2 mg/kg/day of valproic acid, which is 1.2 mg/kg/day above the recommended initial dose range. Her valproic acid level was supratherapeutic when initially checked. Both her initial dose and plasma level exceeded those recommended by the manufacturer (110 mg/ml maximum in women) and might have played a role in her hyperammonemia. However, her maximum dose of 24.3 mg/kg/day was well below the recommended maximum dose of 60 mg/kg/day. Additionally, her encephalopathy persisted after her valproic acid dose was lowered. Her plasma levels appear higher than one might expect for her weight and initial loading doses of valproic acid, and thus an unrecognized variable or risk factor might be influencing them, such as an underlying carnitine deficiency.
Clearly, further study is needed on all potential interactions of valproic acid with psychotropic medications and on other risk factors that might raise valproic acid or ammonia levels. All patients with symptomatic hyperammonemia reported in the psychiatric literature were receiving other medications, psychotropic or otherwise, during treatment.
Treatments
Various treatments for hyperammonemia induced by valproic acid have been tried, including those derived from the treatment of hepatic encephalopathy, such as lactulose or neomycin. Because one way that valproic acid is thought to cause hyperammonemia is by depleting hepatic carnitine, its replenishment might be a treatment specific for encephalopathy induced by valproic acid. According to a consensus panel of pediatric neurologists, there is an association of decreased serum carnitine level with long-term valproic acid administration (36) . However, the panel also recognized that carnitine is mostly stored in muscles and sole reliance on a serum level of carnitine cannot diagnose a true deficiency or a relative deficiency in the liver. Additionally, researchers, such as Coulter (29), have reported cases of encephalopathy with normal serum carnitine levels. Thus, the consensus panel did not feel it necessary to give carnitine to all epileptic children receiving valproic acid therapy, but they strongly recommend it in symptomatic valproic acid-associated hyperammonemia (36) . Specific indications for carnitine use in adults are poorly studied. According to Dr. Raby (personal communication, Aug. 2006), in addition to strict vegetarians, “individuals on restricted diets for medical reasons, especially patients on dialysis” are susceptible to valproic acid’s depletion of hepatic carnitine. Dr. Raby commented further that two meals of lean red meat per week are sufficient to reduce this risk.
Future Directions for Research
An important area for future research is in identifying risk factors for the development of hyperammonemia in adult psychiatric patients. Although the neurological literature is extensive, its overall utility is limited in adult settings because of its focus on children with epilepsy. Areas for study include the role of nutritional status, partial expression of various inborn errors of metabolism, and other medical conditions. Along with risk factors, studies will also need to elucidate drug-drug interactions. Little has been published on the risk of hyperammonemia due to potential interactions of valproic acid with psychotropic medications. Psychiatrists also need to be educated on known drug interactions that increase the risk of hyperammonemia. Unfortunately, the identification of risk factors or drug interactions in severely ill psychiatric patients is often problematic. Because of the presence of psychosis, patients might not have the capacity to understand or answer questions. Medical records are often not available. In such cases, family members who might provide medical history are often unknown to the treating psychiatrist. Any combination of these circumstances makes identification of risk factors difficult.
Diagnosis of hyperammonemia or encephalopathy in psychiatric patients is also an area for study and physician education. After treatment begins, mental status changes due to acute hyperammonemic encephalopathy might be mistaken for improvement in mood, worsening of symptoms of psychosis, or other psychiatric symptoms. Instead of severe symptoms, these might be mild and composed merely of complaints of fatigue. Psychiatrists will need to remain particularly vigilant for delirium when psychosis is present. The distinction between these two illnesses is often difficult to make in a severely ill patient, as our case report indicates. Future studies will need to establish laboratory monitoring recommendations for psychiatric patients taking valproic acid to prevent or identify hyperammonemia. EEG studies might be helpful for identification, and further research is needed to characterize EEG findings in nonepileptic patients with hyperammonemic encephalopathy.
As seen in the case reports, treatments for encephalopathy due to hyperammonemia as a side effect of valproic acid treatment remain empirical and often supportive in nature. Specific treatments have not been tested, making management of this adverse effect an important area for further research. When to restore a possible hepatic carnitine deficiency remains controversial, even within the pediatric neurology population.
With recent evidence of declining results on neuropsychological testing and of neuronal toxicity due to subclinical hyperammonemia in various settings, several authors have expressed concern about the long-term effects of undetected hyperammonemia on cognitive function in patients receiving valproic acid (20, 37) . Authors have recently reported that “chronic hyperammonemia results in cortical and white matter atrophy with Alzheimer type II astrocytes” (38) . Studies are needed to determine, particularly in the psychiatric setting, the prevalence of asymptomatic hyperammonemia among those in long-term treatment with valproic acid, its impact on cognitive function, and what treatments might reduce any impact. Such research is currently underway in pediatric neurology, with an initial study showing no impact of asymptomatic hyperammonemia on cognitive function (39) .
This article has summarized the state of the psychiatric literature on hyperammonemia induced by valproic acid and has placed this psychiatric literature within the context of the studies done in the neurological setting. There are multiple areas where further studies are needed, such as risk factors, prevalence, identification, and treatment. Furthermore, providers in various specialties, including psychiatry, emergency medicine, internal medicine, and even neurology, need education on detecting this reversible form of encephalopathy in patients with psychiatric disorders.
Footnote
Received Jan. 23, 2007; revision received March 19, 2007; accepted April 2, 2007. From the Northern Virginia Mental Health Institute, Falls Church, Va. Address correspondence and reprint requests to Dr. Carr, National Naval Medical Center, Adult Behavioral Health Department, 8901 Rockville Pike, Bethesda, MD 20889-5600; [email protected] (e-mail).Dr. Shrewsbury has received honoraria from Abbott. Dr. Carr reports no competing interests.The views expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Department of the Navy, the Department of Defense, the U.S. government, or the Department of Mental Health, Mental Retardation, and Substance Abuse Services of the Commonwealth of Virginia.The authors thank Dr. J. Nicolai for his review and comments. Dr. Nicolai received no compensation for his contributions.
References
1.
Coulter DL, Allen RJ: Secondary hyperammonemia: a possible mechanism for valproate encephalopathy. Lancet 1980; 1:1310–1311
Kimmel RJ, Irwin SA, Meyer JM: Valproic acid-associated hyperammonemic encephalopathy: a case report from the psychiatric setting. Int Clin Psychopharmacol 2005; 20:57–58
Elgudin L, Hall Y, Schubert D: Ammonia induced encephalopathy from valproic acid in a bipolar patient: case report. Int J Psychiatry Med 2003; 33:91–96
Eze E, Workman M, Donley B: Hyperammonemia and coma developed by a woman treated with valproic acid for affective disorder. Psychiatr Serv 1998; 49:1358–1359
Carlson T, Reynolds CA, Caplan R: Case report: valproic acid and risperidone treatment leading to development of hyperammonemia and mania. J Am Acad Child Adolesc Psychiatry 2007; 46:356–361
Elharmi M, Ferrier B, Martin M, Baverel G: Effect of valproate, sodium 2-propyl-4-pentenoate and sodium 2-propyl-2-pentoate on renal substrate uptake and ammoniagenesis in the rat. J Pharmacol Exp Ther 1993; 266:89–96
Heimberger T: Depakote/Depakene/Depacon (divalproex sodium/valproic acid/valproate sodium): Important Drug Warning, Abbott Pharmaceutical Division; June 2002 Safety Alerts for Drugs, Biologics, Medical Devices, and Dietary Supplements, Food and Drug Administration. www.fda.gov/medwatch/SAFETY/2002/depakote_deardoc.pdf
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
For more information or tips please see 'Downloading to a citation manager' in the Help menu.
PsychiatryOnline subscription options offer access to the DSM-5-TR® library, books, journals, CME, and patient resources. This all-in-one virtual library provides psychiatrists and mental health professionals with key resources for diagnosis, treatment, research, and professional development.
Need more help? PsychiatryOnline Customer Service may be reached by emailing [email protected] or by calling 800-368-5777 (in the U.S.) or 703-907-7322 (outside the U.S.).
If the address matches an existing account you will receive an email with instructions to retrieve your username
Create a new account
Change Password
Password Changed Successfully
Your password has been changed
Login
Reset password
Can't sign in? Forgot your password?
Enter your email address below and we will send you the reset instructions
If the address matches an existing account you will receive an email with instructions to reset your password.
Change Password
Congrats!
Your Phone has been verified
×
As described within the American Psychiatric Association (APA)'s Privacy Policy and Terms of Use, this website utilizes cookies, including for the purpose of offering an optimal online experience and services tailored to your preferences. Please read the entire Privacy Policy and Terms of Use. By closing this message, browsing this website, continuing the navigation, or otherwise continuing to use the APA's websites, you confirm that you understand and accept the terms of the Privacy Policy and Terms of Use, including the utilization of cookies.