Neurotransmitters, the key information molecules of the brain, mediate the actions of all known psychoactive drugs. We currently know of at least 50 to 100 possible neurotransmitters representing diverse chemical classes, including the biogenic amines, amino acids, peptides, and gases. As recently as the late 1950s, we knew of only two neurotransmitters, acetylcholine and norepinephrine. Acetylcholine was well established as the neurotransmitter that is active at the neuromuscular junction and in the autonomic nervous system. Norepinephrine was appreciated as the neurotransmitter of postganglionic sympathetic neurons. It was assumed that acetylcholine was important in the brain, but its functions in the central nervous system (CNS) were unclear. Norepinephrine had not yet been identified in CNS neurons. During these years, seminal observations were spawning the field of psychopharmacology. Newly developed flourometric techniques permitted the simple and specific measurement of biogenic amines. Soon it was discovered that reserpine, which in the late 1950s had been associated with depression in some patients, depleted the brain of serotonin and norepinephrine, whereas monoamine oxidase inhibitors, which had antidepressant effects, elevated the levels of these biogenic amines. There emerged a simplistic but reasonably accurate idea that norepinephrine and serotonin were important determinants of mood, with low levels causing and elevated levels reversing depression.
What was extraordinary about this early work, besides its prescience, was that this functional/psychiatric insight about biogenic amines preceded any evidence that these chemicals were neurotransmitters or even that they were contained within neurons. Establishing the presence of a substance in neurons and visualizing the specific neuronal populations containing the substance provides a quantum leap in insight. For biogenic amines, this information did not emerge until the mid 1960s, when a histochemical technique for visualizing the amines, developed by the Swedish investigator Nils-Erik Hillarp, was applied to the brain by his students Kjell Fuxe and Annica Dahlstrom. They successfully mapped out the biogenic amine neuronal pathways in the brain
(1).
These historical reflections highlight a long-standing question in the neurosciences that is not yet fully resolved: what is a neurotransmitter? Over the years, criteria for defining a neurotransmitter have become so problematic that many neuroscientists gloss over the problem by labeling substances with vague words such as “neuromodulator.” For purposes of classifying neurotransmitters, neurophysiologists have discriminated two principal types of physiologic actions, fast and slow. Fast effects usually involve opening or closing of ion channels that are often part of a receptor protein, such as the sodium channel that is contained within the nicotinic acetylcholine receptor. Slow actions typically arise from metabolic alterations in target cells whose receptor proteins are termed metabotropic. Classic examples include the formation of cyclic GMP, cyclic AMP, or inositol trisphosphate by enzymes that are linked to receptors via GTP binding G-proteins (e.g., the muscarinic receptors for acetylcholine). Neurotransmitters are believed to signal through both fast and slow mechanisms. What else defines a neurotransmitter?
Although most of us have a vague idea of what we expect of a neurotransmitter, some neuroscientists have enunciated concrete criteria for “transmitterhood.” Since acetylcholine was the first neurotransmitter to be identified and characterized, the early criteria assumed that candidate neurotransmitters should be “just like” acetylcholine. For example, it was known that acetylcholine is synthesized by choline acetyltransferase, stored in synaptic vesicles, and released into the synapse after fusion of the vesicles with the plasma membrane in response to elevated calcium through exocytosis. Thus, a candidate transmitter should be localized to neurons, have a special biosynthetic enzyme, and be released in a calcium-dependent fashion when the nerve is depolarized. Of course, the candidate transmitter should mimic the actions of the endogenous transmitter when applied to postsynaptic cells. Acetylcholine, when acting postsynaptically, binds to specialized receptor proteins such as the nicotinic and muscarinic acetylcholine receptors. Accordingly, any new candidate transmitter was also expected to bind to a receptor protein localized to the external surface of the target cell’s plasma membrane. Finally, to mimic acetylcholine, an enzyme near the receptor (i.e., like acetylcholinesterase) should degrade and thereby inactivate the transmitter at the synapse.
Thus, a neurotransmitter has been thought of as a chemical stored in a nerve terminal that is released when the nerve fires to act on adjacent cells, altering their level of excitation. However, there are many challenges to this straightforward conceptualization. For example, neurophysiologists have now identified many unconventional ways that neurons can signal to each other via their cell bodies, dendrites, and portions of the axons other than the nerve terminals. These findings have generated new questions about neurotransmitter functions. Might a neurotransmitter be released from a dendrite rather than a nerve terminal? Can a chemical that acts on the same neuron that released it be considered a neurotransmitter? Neurons account for only a minority of cells in the brain, with glia making up about 85% of total. Although originally characterized as “supporting cells,” glia are now known to display a number of interesting electrical and chemical features. Might a substance that acts on glial cells rather than neurons be considered a neurotransmitter? Can glia release neurotransmitters?
By the early 1960s, difficulties with the acetylcholine-based definition of a neurotransmitter became evident as research moved beyond acetylcholine. In the early characterization of norepinephrine, monoamine oxidase, the only known degrading enzyme, was presumed to provide synaptic inactivation. However, monoamine oxidase inhibitors did not potentiate sympathetic neurotransmission. When Julius Axelrod discovered catechol-
O-methyltransferase (COMT), he thought that this enzyme might constitute the synaptic inactivating system. However, COMT inhibitors also failed to potentiate synaptic transmission. Axelrod
(2) and his associates then made a key discovery: synaptic inactivation of norepinephrine did not involve enzymes at all but rather a novel reuptake mechanism whereby the presynaptic neuron removed the neurotransmitter from the synapse by transporting it back into the nerve that had released it. If one applied the original, rigid criteria for a transmitter based on the characteristics of acetylcholine, norepinephrine would fail to qualify. Over time, it became evident that reuptake is common. Almost all biogenic amines, including serotonin and dopamine, are inactivated by reuptake
(3,
4). Histamine provides an interesting exception to this generalization. Although histamine is formed by a specific enzyme, histidine decarboxylase, and is localized to discrete neuronal populations in the brain, there is no evidence that reuptake accounts for its synaptic inactivation. In addition, the histamine metabolizing enzymes, histamine methyltransferase and diamine oxidase, do not account for histamine’s inactivation. Thus, the question of whether specific synaptic inactivation terminates histamine signaling remains unanswered
(5).
In addition to biogenic amines, amino acids were found to have neurotransmitter functions. Signaling by γ-aminobutyric acid (GABA), glutamate, glycine, and other amino acid transmitters, like signaling by the biogenic amines, is terminated by specific reuptake transport proteins. Gradually, neuroscientists accepted that neurotransmitter reuptake is as valid a means of providing synaptic inactivation as enzymatic degradation. In fact, reuptake eventually became appreciated as the rule rather than the exception.
The discovery and characterization of amino acid transmitters in the brain challenged other dogma about what constitutes a neurotransmitter. GABA was readily accepted as a transmitter because it is primarily, if not exclusively, devoted to a neurotransmitter role, and GABA is synthesized by a specific enzyme, glutamic acid decarboxylase. Moreover, the enzyme is localized to discrete neurons, GABA neurons. Glutamate and glycine were far more difficult for the scientific community to accept, since these amino acids are involved in protein synthesis and other metabolic pathways, with only a minor fraction of the total neuronal pool involved in a transmitter role. Glutamate posed a particular challenge, because its total concentration in the brain is extraordinarily high (≈20 mM), and glutamate is crucial in numerous pathways of intermediary metabolism. Even now, it is difficult to identify histochemically exactly which neurons in the brain employ glutamate as a neurotransmitter. Since one cannot identify specific biosynthetic enzymes for glutamate and glycine, one could argue that these molecules do not satisfy classic criteria for a neurotransmitter.
Despite such initial concerns, neuroscientists now accept amino acids as neurotransmitters with widespread functions in the brain. Although exact percentages are hard to pin down, glutamate is likely the neurotransmitter for 50% or more of synapses in the brain and is unquestionably the principal excitatory neurotransmitter. Aspartate chemically resembles glutamate, having just one less methyl group, so the two are difficult to distinguish in terms of transmitter function and localization. However, aspartate is thought to be an important excitatory neurotransmitter in the spinal cord. GABA is the principal inhibitory neurotransmitter in the brain and occupies 25%–40% of synapses, depending on the brain region. In the spinal cord, glycine may be the major inhibitory transmitter, signaling in 25%–30% of synapses. By contrast, dopamine, norepinephrine, and serotonin each are thought to account for only about 1% of brain synapses, and acetylcholine might occupy up to 5%. Thus, amino acids are quantitatively the principal neurotransmitters in the brain.
Like the amino acids, adenosine is involved in multiple metabolic functions and occurs in high concentrations in all tissues. Adenosine is neither a biogenic amine nor an amino acid, although it does possess amine-like properties. Discrete neuronal populations, with uniquely high densities of adenosine, have been identified by immunohistochemistry, suggesting a transmitter role
(6). Adenosine is neuroactive, acting on nerve terminals to inhibit the release of most neurotransmitters. Moreover, a robust reuptake system for adenosine exists, and potent, selective adenosine reuptake inhibitors potentiate adenosine effects on neural activity.
By the end of the 1960s, acetylcholine, biogenic amines, and amino acids had been generally accepted as neurotransmitters. The 1970s became the decade of the peptide transmitters
(7). Substance P was the first appreciated neuropeptide. It was discovered in the 1930s as an unidentified factor in tissue extracts that caused smooth muscle contraction. Substance P was isolated and sequenced by Susan Leeman
(8), culminating her efforts to identify a tissue component that stimulates salivation in rats. Widespread interest in neuropeptides followed the identification of opiate receptors and the discovery of enkephalins as their endogenous ligands
(9,
10). Enkephalins are small peptides that contain five amino acids. They are highly localized to the same discrete sites as opiate receptors. Actions of the enkephalins closely mimic those of morphine, which led to rapid acceptance of the enkephalins as neuroactive substances with physiologic roles in the brain. The localization of the enkephalins made use of immunohistochemistry, a technology pioneered through the elegant studies of Tomas Hokfelt and his associates
(11), as a tool to characterize neuropeptides. In the late 1970s and early 1980s immunohisochemistry contributed to a veritable explosion in the number of identified neuropeptides. During this time, substance P was found to be enriched in intestinal neurons as well as brain neurons. Indeed, many neuropeptides were first identified in the intestine and related organs such as the pancreas. Examples include vasoactive intestinal polypeptide, cholecystokinin, gastrin, and even insulin
(12).
During the time neuropeptides were being discovered in the brain and intestine, one might have argued that the amines and amino acids had “filled up the brain” so that no neuronal populations without transmitters remained. The question of which neurons would use neuropeptides was addressed by Hokfelt and associates
(11), who established evidence for cotransmitters. We now know that most neurons contain an amino acid transmitter stored together with a biogenic amine or a peptide. Although any given neuropeptide probably accounts for only 1% or fewer of synapses, the 50 to 100 known neuropeptides may collectively occupy a substantial portion of CNS neurons. It is now generally accepted that that few if any brain neurons contain a single transmitter.
Neuropeptides also challenge the dogmatic criteria for a neurotransmitter. In terms of synaptic inactivation, peptides are presumably hydrolyzed by various peptidases. However, no one has rigorously demonstrated that a specific peptidase accounts for synaptic activation of individual neuropeptides. Most peptides do not elicit neuronal excitation or inhibition, as do acetylcholine and GABA. Rather, neuropeptides appear to have a modulatory action that has been difficult for neurophysiologists to characterize clearly. In some instances, certain actions of serotonin, norepinephrine, and dopamine also appear modulatory. This characteristic has led some to deny these molecules “transmitter status” and relegate them to the muddled role of neuromodulators. Perhaps, if the discovery of neuropeptides had preceded acetylcholine, the latter would have been challenged to fulfill transmitter criteria modeled on neuropeptide function.
By the mid 1980s so many neuropeptides had been identified that most neuroscientists were ready to close the book on chemical classes of neurotransmitters, with completed “chapters” on biogenic amines, amino acids, and neuropeptides. The 1990s have brought us two new major chemical classes of novel neurotransmitters that we will now explore in detail: gases and d-amino acids.
Gases
Nitric Oxide
The physiological role of nitric oxide (NO) was discovered in the 1980s in studies of blood vessels and macrophage activity. In 1980, Robert Furchgott discovered that the ability of acetylcholine to relax blood vessels did not involve direct actions through cholinergic receptors on smooth muscle
(13). Instead, acetylcholine’s action required the endothelium to elaborate a vasoactive substance that would enter the smooth muscle to relax the muscle. This endothelial derived relaxing factor (EDRF) was later shown to be NO
(14–
16). Independently, other workers were trying to explain how activated macrophages kill tumor cells and bacteria. Arginine, later found to be the precursor of NO, was known to be crucial, and NO was identified as the key active molecule
(15,
17–
19). Garthwaite and colleagues
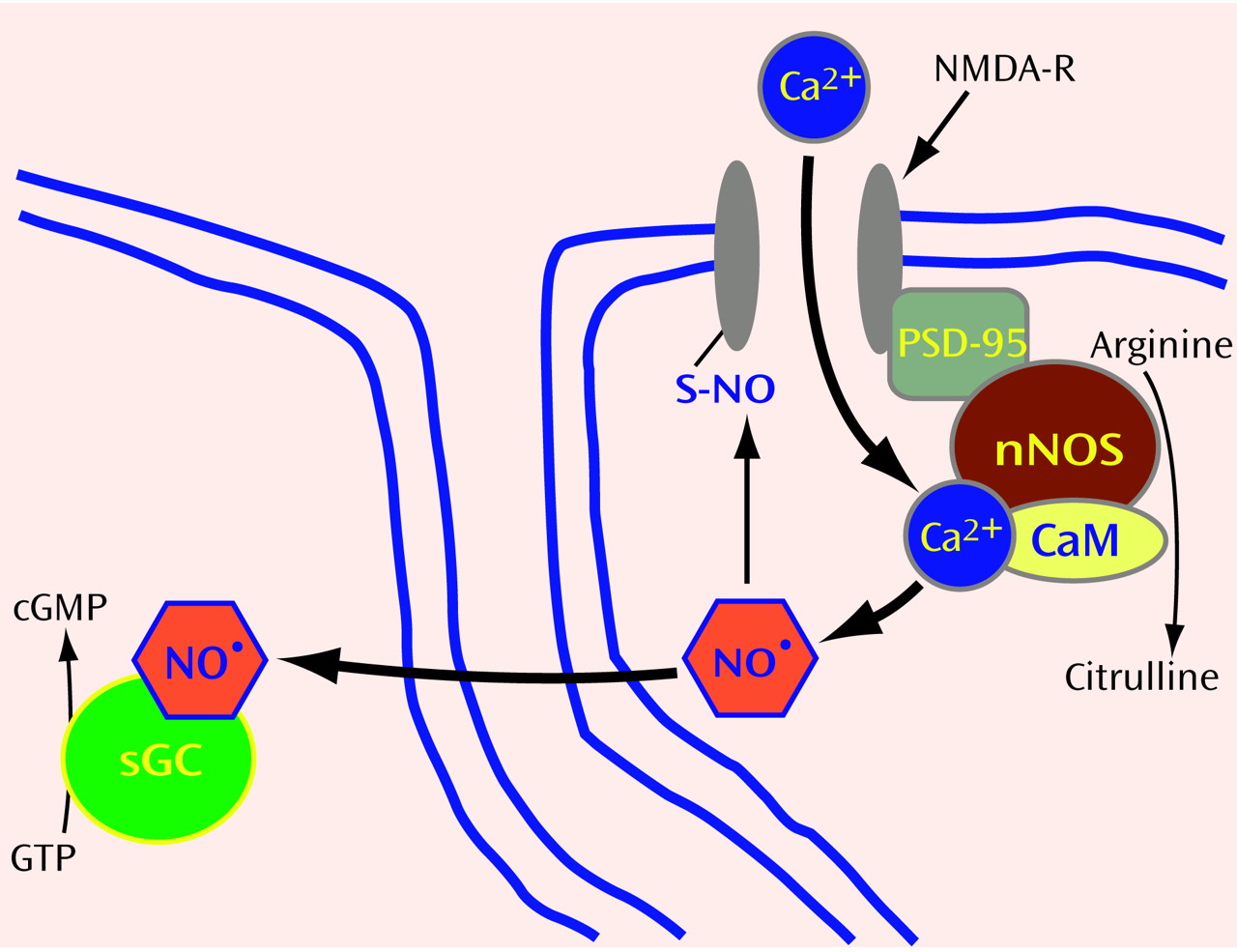
(20) reported evidence for a substance with EDRF activity in the cerebellum. We became curious about a possible role for NO in the brain. Since it was virtually impossible at that time to measure NO gas directly, we decided to focus on the enzyme that makes NO. NO is generated in a single step from the amino acid arginine through the action of an enzyme designated NO synthase (NOS) (
Figure 1 and
Figure 2). Using NADPH as an electron donor, NOS oxidizes one of the guanidino nitrogens of arginine to form NO, with citrulline as an amino acid coproduct. Despite extensive investigations, a neuroactive role for citrulline has not been found.
To establish whether NO plays a role in neurotransmission in the brain, we took advantage of what was already known about how NO relaxes smooth muscle in blood vessels. By stimulating the activity of soluble guanylyl cyclase, the enzyme that makes cyclic GMP, NO initiates a signaling cascade that involves protein phosphorylation and causes smooth muscle relaxation. The cerebellum contains the highest levels of cyclic GMP in the brain. In addition, it was known that glutamate could trigger a 10-fold augmentation of cyclic GMP levels through activation of the
N-methyl-
d-aspartate (NMDA) subtype of glutamate receptors. As an index of NOS activity, we monitored the conversion of radiolabeled arginine to citrulline in ex vivo preparations of rat cerebellum. After NMDA receptor activation, NOS activity rapidly tripled in parallel with increased cyclic GMP levels
(22), an effect also noted by Garthwaite and colleagues
(23). We used arginine derivatives that inhibit NOS activity by competing with arginine for the active site in NOS to explore a link between NOS and cyclic GMP. NOS inhibitors specifically blocked the rise in cyclic GMP. Therefore, we could attribute increased cyclic GMP production to NO, establishing a role for NO in mediating actions of glutamate in the brain.
Clearly, NO dynamics are determined by NOS activity. Thus, it was crucial to characterize this enzyme. Several groups had tried to purify the NO-generating protein without success, because most purification processes led to loss of enzyme activity. We guessed that the enzyme itself might not be particularly labile, but that a cofactor might be dissociated during the purification. Knowing that calcium augmented enzyme activity, we hypothesized that the calcium-binding protein, calmodulin, might be the cofactor. When calmodulin was added to partially purified preparations, NOS activity was completely restored
(24). Besides permitting purification of the enzyme, this finding explained how glutamate, through NMDA receptor activation, could almost immediately stimulate NO formation: after the NMDA receptor ion channel opens in response to binding glutamate, calcium enters the neuron, where it binds calmodulin to form a calcium-calmodulin complex (CaM) that binds and activates NOS.
Subsequently, we biochemically purified NOS
(24) and developed specific antibodies to allow immunohistochemical visualization of NO neurons
(25). NOS-containing neurons have very discrete localizations and represent only about 1% of neuronal cells in the brain. However, their axons ramify so extensively that virtually every cell in the brain may encounter a NOS nerve terminal
(26,
27). Presumably, glutamate neurons synapse on NOS neurons, enabling NMDA receptor activation to trigger NO formation. As a diatomic gas, NO is freely diffusible and thus can readily enter adjacent neuronal cells or other cells (
Figure 1). Once inside target cells, NO binds the iron in heme contained within the active site of soluble guanylyl cyclase, activating the enzyme to form cyclic GMP.
Given what we know about the synthesis and action of NO, does NO fulfill classic criteria for a neurotransmitter? In the brain, NO is formed in neurons in response to calcium influx, reminiscent of calcium dependent exocytic release of other neurotransmitters. However, as a gas, NO cannot be stored in synaptic vesicles or released by exocytosis. Moreover, there are no “receptors” for NO on the postsynaptic membrane of adjacent cells. Instead, NO diffuses from NOS neurons into neighboring cells, where it binds guanylyl cyclase, an “enzyme receptor.”
For most neurotransmitters, only a small percentage of the total store is released with each nerve stimulation, as there is a large storage pool of neurotransmitter in synaptic vesicles. Since NO cannot be stored, NOS must be activated every time a neuron needs to release NO. Accordingly, one might expect NOS enzymatic activity to be exquisitely regulated. Biochemical purification of NOS allowed us to clone the gene encoding it, obtain the full amino acid sequence, and express the recombinant protein in vitro for detailed characterization
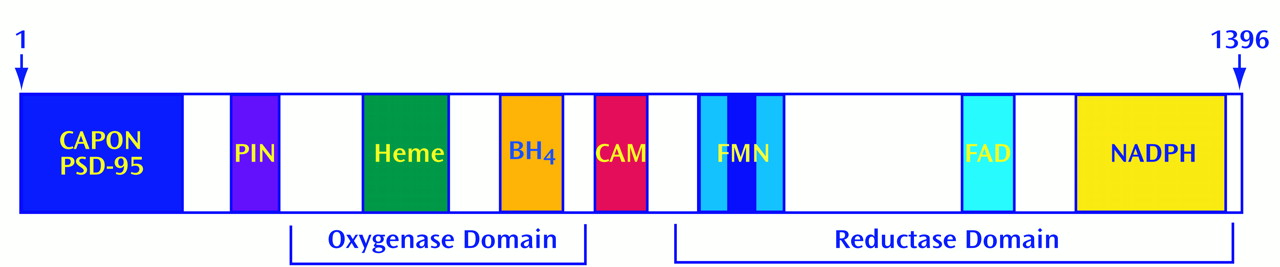
(27). NOS contains multiple sites for regulation
(28). Although most oxidative enzymes use one electron donor, NOS is more complex. NOS possesses tightly bound flavin adenine mononucleotide (FMN) and flavin adenine dinucleotide (FAD), in addition to NADPH. It also utilizes heme and tetrahydrobiopterin (BH
4) as electron donors. NOS possesses sites for phosphorylation by the major phosphorylating enzymes, including cyclic AMP-dependent protein kinase (PKA), protein kinase C (PKC), calcium-calmodulin-dependent protein kinase (CaMK), and cyclic GMP-dependent protein kinase (PKG)
(28–
30). Recently, NOS in endothelial cells has been shown to be phosphorylated by protein kinase B (PKB), also known as Akt, which participates in signaling cascades that affect nuclear function
(31–
33).
NOS can be regulated by interactions with other proteins. In the brain, NOS is physically linked to the postsynaptic membrane near NMDA receptors through its interaction with postsynaptic density protein-95
(34). By using the yeast 2-hybrid technique, we identified two other NOS binding partners: a protein inhibitor of NOS (PIN)
(35) and carboxyl-terminal PDZ ligand of NOS (CAPON)
(36). PIN is a phylogenetically conserved small protein that inhibits neuronal NOS (nNOS) by preventing the formation of NOS dimers, the form required for enzyme activity. CAPON appears to be a chaperone or scaffolding protein that links nNOS to other proteins.
Molecular cloning of NOS from brain permitted the identification of genes for distinct NOS isozymes from endothelial cells in blood vessels
(37–
39) and macrophages
(40–
42). The form of NOS that we initially purified and cloned was designated nNOS (neuronal NOS), the macrophage form is termed inducible NOS (iNOS), and the endothelial form is called endothelial NOS (eNOS). iNOS is so designated because, under resting conditions, macrophages and other cells display negligible enzyme activity. However, in response to physiologic stimuli, such as exposure to lipopolysaccharide from Gram-negative bacteria, these cells are induced to express iNOS and generate large amounts of NO sufficient to kill bacteria or tumor cells. Such stimuli provoke new synthesis of iNOS enzyme protein in just 1–2 hours. By contrast, nNOS and eNOS proteins are constitutively present and, as described above for nNOS, are activated by CaM.
Functions of NO
Experimentally establishing that a substance is a neurotransmitter in the brain can be exceedingly difficult. The intestine provides an excellent model system for the study of neurotransmitters since smooth muscle function can be used to monitor neurotransmitter release from the enteric nervous system, and the enteric nervous system uses nearly all transmitters found in the brain. Indeed, the most direct evidence for NO as a neurotransmitter comes from studies of the intestine, in which many neurons express nNOS. The excitatory transmitters in the enteric nervous system, including acetylcholine, norepinephrine, and some neuropeptides, have been known for some time. In contrast, the identity of the major inhibitory transmitters has been controversial. Since inhibitory transmission persists in the presence of selective blockade of adrenergic and cholinergic receptors, inhibitory transmission was designated nonadrenergic, noncholinergic (NANC) transmission. Stimulation of intestinal neurons causes the elaboration of a vasorelaxant factor that is indistinguishable from NO
(43). NOS inhibitors block NANC transmission
(44–
46). To further clarify the role of NO as a neurotransmitter, mice with targeted genomic deletion of the nNOS gene (nNOS knockout mice) were developed
(47). In small intestinal smooth muscle preparations from these mice, we found that NANC transmission was reduced by about 50%
(48). More recently, we have found that smooth muscle cells from nNOS knockout mice have abnormal resting membrane potentials, implying that basal NO production determines the excitability of intestinal smooth muscle
(49). Moreover, compounds that generate NO, when added to intestinal preparations, can mimic NANC transmission. Taken together, these findings establish a role for NO as a neurotransmitter.
We also found nNOS localized to neurons that innervate the corpora cavernosae and blood vessels of the penis
(50). Electrical stimulation of the cavernous nerves provokes erection, which is blocked by NOS inhibitors. These results establish NO as a neurotransmitter mediating penile erection. As in other organs, NO mediates erection through the stimulation of cyclic GMP formation. Sildenafil inhibits phosphodiesterase type 5, an enzyme that selectively degrades cyclic GMP
(51). Thus, the therapeutic effect of sildenafil for patients with erectile dysfunction involves potentiation of NO neurotransmission. It is of interest that sildenafil has no demonstrable effects on mental function, aside from eliciting mild headaches, presumably by causing vasodilatation of scalp blood vessels. This may relate to the limited ability of the drug to penetrate the blood-brain barrier.
Mice lacking nNOS and therefore NO production can be studied to ascertain physiologic roles for neuronally derived NO. Gross anatomic observations have revealed only a greatly dilated stomach with hypertrophy of the pylorus
(47). Recently, we found that NANC relaxation of the pyloric sphincter is abolished in nNOS knockout mice and restored by NO donors
(52). Since the pylorus contains a plexus of nNOS neurons, NO appears to be the neurotransmitter mediating NANC relaxation of the pylorus. This finding has clinical relevance, as nNOS knockout mice appear to be an animal model of infantile hypertrophic pyloric stenosis. In patients with this condition, nNOS protein cannot be detected in the pylorus, and the pyloric muscle is hypertrophied as in the nNOS knockout mice
(53,
54). Since the gastric dilation and pyloric dysfunction of the nNOS knockout mice resembles the gastric dysfunction observed in many diabetic patients, we reasoned that the nNOS knockout mice might also represent an animal model of diabetic gastropathy
(55–
57). In diabetic mice, we found that NO-dependent NANC transmission is lost in the pyloric muscle
(52). At the same time, nNOS protein and mRNA are absent from the pyloric neurons, although the nerves themselves remain intact. Insulin treatment restores nNOS protein and mRNA expression in conjunction with a return of NO-mediated NANC pyloric relaxation. These results are consistent with a regulatory effect of insulin or glucose on nNOS expression, perhaps through regulatory sites in the promoter of the nNOS gene
(58–
60).
Insight into a physiologic role for NO in the brain comes from behavioral studies of nNOS knockout mice. These mice are extraordinarily aggressive, more so than virtually any other form of genetically mutant mice previously described
(61). This behavior is dependent on testosterone, as it is abolished by castration, reproduced by testosterone administration, and absent in females
(62). nNOS knockout male mice also display abnormal, excessive sexual activity. When a normal male mouse is placed together with a female that is not in estrus (the sexual receptive phase of the estrous cycle), the male will begin to mount the female but will cease when he perceives that the female is not responsive. Male nNOS knockout mice fail to heed such clues and will repeatedly mount the female
(61). These findings imply that in males nNOS neurons normally restrain aggressive and sexual behaviors. In contrast, in females, maternal aggression is reduced in nNOS deficient mice
(63).
NO may also play a role in learning and memory. Long-term potentiation is a model of synaptic plasticity in which powerful stimulation of a synaptic input to a neuronal system potentiates subsequent synaptic transmission in the system for long periods of time. In intact animals, long-term potentiation can persist for weeks. Chemicals that release NO facilitate long-term potentiation. There has been controversy about possible decreased long-term potentiation in nNOS-deficient mice, perhaps because of compensation by related genes like eNOS. Elegant studies by Kandel and associates show a clear decrease of long-term potentiation in mice with deletion of both eNOS and nNOS
(64). Whether eNOS is located in neurons or only blood vessels in the brain remains controversial. Perhaps, NO produced in blood vessels can influence neural transmission. This possibility fits with evidence for behavioral changes in eNOS knockout mice. These mice display a pronounced decrease in aggressive behavior, opposite to the nNOS knockout mice
(65). This result may seem surprising, but localizations of eNOS associated with brain vasculature differ markedly from those for nNOS, thus NO formed by the two different enzymes may influence distinct and largely unrelated brain structures.
NO dysfunction in stroke
As medical students, many of us were taught that stroke reflects permanent damage in infarcted tissue where cerebral arteries have been occluded. However, evidence accumulated over the last decade has indicated that a major fraction of the neural damage in stroke is due to oxygen-derived free radicals that are produced after reperfusion of the ischemic area or from hypoxic mitochondria in surviving neurons. We also know that hypoxia in the brain triggers a massive release of excitatory transmitters, especially glutamate. Glutamate levels may reach as high as 50 times normal levels, literally “exciting to death” partially hypoxic cells
(66). Evidence for a role of glutamate in stroke includes the ability of glutamate antagonists, especially NMDA receptor antagonists, to reduce stroke damage about 50%–60%
(66). Glutamate toxicity associated with stroke can be mimicked in cerebral cortical cultures where NMDA receptor activation can kill up to 90% of neurons, while NMDA receptor antagonists prevent this damage
(67). Glutamate neurotoxicity is diminished in cultures from nNOS knockout mice or after treatment with NOS inhibitors
(68). Stroke damage is also markedly reduced after treatment with NOS inhibitors
(69–
73) and in nNOS knockout mice
(74).
If NO mediates neurotoxicity in stroke, how exactly does NO kill cells? Although NO is a free radical, it is not a particularly toxic one. When NO combines with superoxide (e.g., from hypoxic mitochondria), it forms peroxynitrite, which degenerates into the extremely toxic hydroxyl radical (OH&*;. Peroxynitrite and OH&*; can damage all major biomolecules, including lipids (through peroxidation of cell membranes leading to calcium entry), proteins (both directly and through calcium-dependent proteases), and DNA. All of these processes may play some role in neurotoxicity, but some evidence suggests that DNA damage is the major mechanism. OH&*; causes DNA strand breaks that activate the enzyme poly (ADP-ribose) polymerase (PARP). PARP is a nuclear enzyme that facilitates the DNA repair process. PARP’s substrate is NAD, which, when activated, transfers 50 to 200 ADP-ribose groups in branched chains to several nuclear proteins, including PARP itself. When NAD is overactivated by massive amounts of DNA damage, NAD levels are depleted, and ATP is also depleted in efforts to resynthesize NAD
(75,
76). Accordingly, DNA damage leading to PARP overactivation can kill cells by energy loss and cellular starvation. Evidence that such a mechanism mediates killing of cells by NO comes from studies showing that PARP inhibitors block neuronal death in cultures elicited by NO donors or NMDA receptor activation
(77). Even more strikingly, brain cultures from PARP knockout mice are completely protected from such neurotoxicity
(78). PARP-mediated neuronal cell death may have clinical relevance. In PARP knockout mice, there is a 80% reduction in stroke damage after reversible occlusion of the middle cerebral artery
(78,
79), and postischemic administration of a PARP inhibitor can ameliorate stroke damage
(80).
Carbon Monoxide
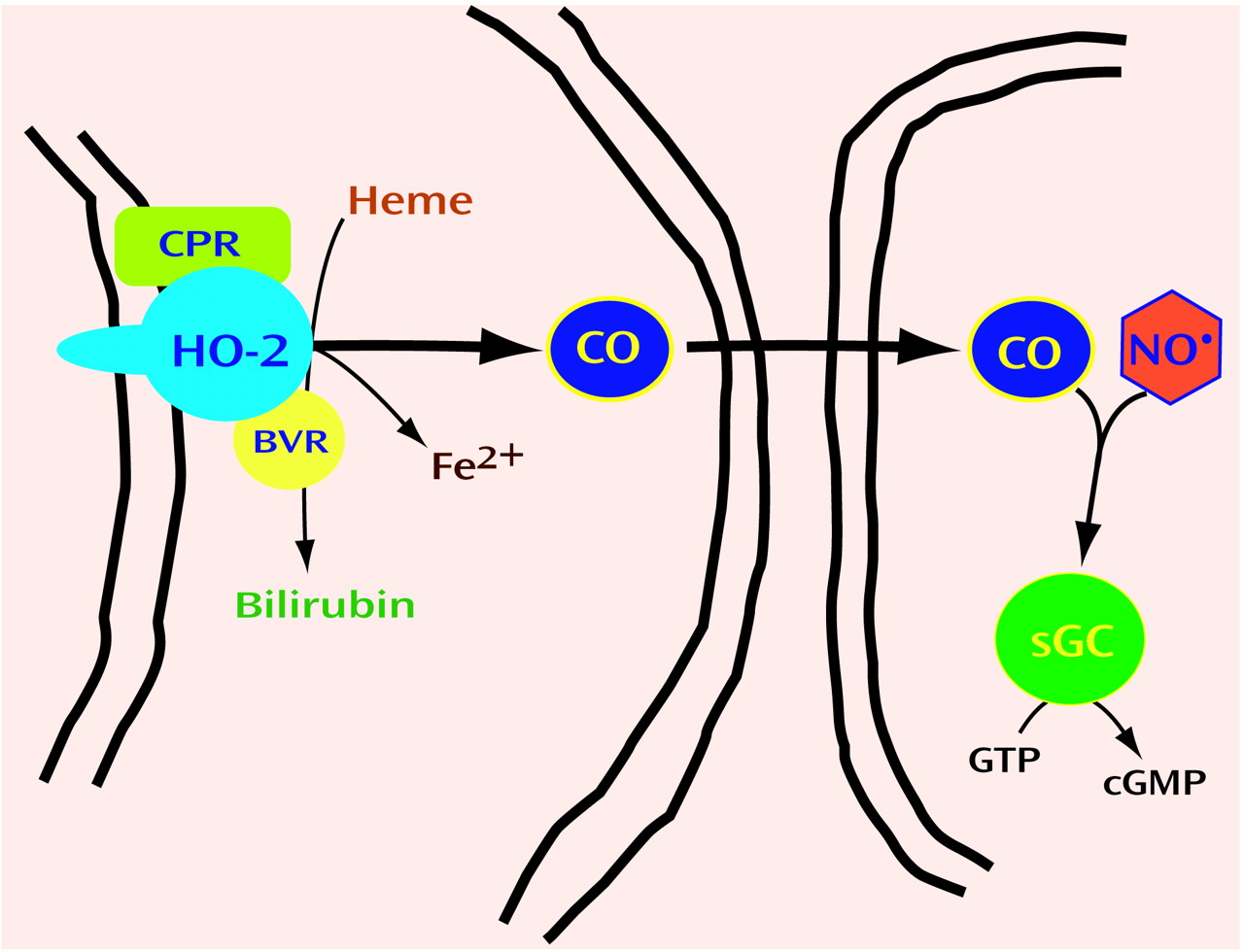
Neurotransmitters are grouped by chemical classes, such as the biogenic amines, amino acids, and neuropeptides. Thus, we wondered whether gaseous transmitters besides NO might exist. Carbon monoxide (CO) is formed physiologically by heme oxygenase (HO). HO cleaves the porphyrin ring of heme to form biliverdin, which is rapidly reduced by biliverdin reductase to bilirubin (
Figure 3). In the process, ferrous iron is released, and a one-carbon fragment is released as CO. Might CO be the second member of a new group of neurotransmitters, the gases?
HO was first identified as an inducible enzyme activated by many cellular stresses. This inducible form of HO was found to be identical to a heat shock protein and is now designated HO1. HO1 is most concentrated in the spleen, the repository of aged red blood cells. In the spleen, HO degrades heme from red blood cell hemoglobin which otherwise would be quite toxic. In other cells, HO destroys heme from mitochondrial and other enzymes. In the course of purifying HO1, Maines and associates
(85,
86) discovered a second HO protein, which they designated HO2. HO2 is not inducible and is most concentrated in the brain and testes. We reasoned that if CO were to be a neurotransmitter, its biosynthetic enzyme should be discretely localized to certain neurons. In situ hybridization studies revealed that HO2 mRNA is highly localized to specific neurons in the brain, with localizations similar to those of soluble guanylyl cyclase
(87). Like NO, CO can activate this enzyme, triggering cyclic GMP formation (
Figure 3). In cultures of olfactory neurons, which have very high concentrations of HO2 and lack nNOS, cyclic GMP levels are depleted by HO inhibitors
(88,
89).
As in the case of NO, direct evidence for CO as a neurotransmitter emerged from studies of the myenteric plexus of the intestine. Immunohistochemical staining revealed significant colocalization of HO2 and nNOS in myenteric neuronal cells, suggesting that CO and NO might be cotransmitters
(48,
90). As discussed above, NANC transmission is reduced by half in intestinal preparations from nNOS knockout mice. In similar experiments, we used mice with targeted genomic deletion of HO2 (HO2 knockout mice) and found that NANC transmission was also reduced 50% in intestine in those mice
(48). Recently, we have extended these findings by demonstrating depolarization of the resting membrane potential in jejunal smooth muscle cells from HO2 knockout mice
(49). Intestinal preparations from these mice also have decreased NANC inhibitory junctional potentials, measured electrophysiologically. In mice deficient in both nNOS and HO2, we observed additive effects on the resting membrane potential and the inhibitory junctional potentials
(49). Application of exogenous CO mimics NANC neurotransmission. Thus, CO, like NO, appears to be an inhibitory NANC neurotransmitter.
HO2 is also concentrated in nerves that innervate the vas deferens, a smooth muscle whose contractions underlie ejaculation. Reflex activity of the bulbospongiosus muscle that underlies ejaculation is profoundly reduced in preparations from HO2 knockout mice, and the mice display a marked reduction in ejaculatory function
(91). Bulbospongiosus nerves do not contain nNOS. Conversely, the NO nerves mediating penile erection do not contain HO2. Although NO is critical for penile erections, CO appears to be the neurotransmitter of nerves mediating ejaculation. Thus, in the myenteric plexus, NO and CO may be cotransmitters, but in other peripheral autonomic nerves, they have distinct localizations and functions.
As described above, CO appears to physiologically regulate cyclic GMP in olfactory neurons. However, its functional link to cyclic GMP in other parts of the brain is not clear despite the apparent colocalization of HO2 and soluble guanylate cyclase. Although some relatively nonspecific inhibitors of HO diminish long-term potentiation, no major deficit in long-term potentiation is evident in HO2 knockout mice
(92–
94). Thus, a transmitter role for CO in the brain remains to be rigorously established. Even signaling through cyclic GMP (cGMP) in the brain is not established, as whole brain levels of cGMP are not reduced in HO2 knockout mice (R. Zakhary and S.H. Snyder, unpublished 1999 data). Although the knockout animals are useful research tools, drugs that selectively perturb transmitter actions are comparably important. Whereas potent, selective inhibitors of NOS exist, the existing HO inhibitors, mostly protoporphyrin structures, are only modestly selective for HO.
In contrast to the current paucity of evidence regarding a role for CO in normal brain function, other products of HO enzymatic activity do display physiologic roles. Ames and co-workers identified an antioxidant function for bilirubin many years ago
(95,
96). Recently, we showed a neuroprotectant role for bilirubin
(82). This work stemmed from our efforts to identify a means for activating HO2 analogous to the CaM activation of nNOS. Phorbol esters, which stimulate protein kinase C, protect cortical neurons from death after treatment with hydrogen peroxide. The neuroprotection of phorbol esters is greatly reduced by inhibitors of HO and is abolished in neuronal cultures from HO2 knockout mice. Bilirubin, in very low concentrations, mimics the neuroprotective effects of phorbol esters. In findings consistent with a neuroprotective role for bilirubin, stroke damage was substantially worsened in HO2 knockout mice
(83). Sometimes, mice with a specific genetic deficiency are unhealthy and therefore susceptible to injury or illness in general. HO1 knockout mice are generally debilitated, with weight loss, anemia, and signs of chronic inflammation developing after 20 weeks of age. However, HO1 knockout mice do not display accentuated stroke damage. In contrast, HO2 knockout mice appear normal and healthy, though they suffer enlarged strokes in ischemic models.
The neuroprotective effects of bilirubin are surprising considering the well-known neurotoxicity of bilirubin in jaundiced babies with very high blood levels of bilirubin. Kernicterus requires high micromolar levels of bilirubin
(97), a thousand times the concentrations of bilirubin that are neuroprotective in vitro. There is interesting evidence that the moderately “elevated” plasma levels of bilirubin in most normal babies may be protective, as babies with moderately elevated bilirubin levels are less susceptible to oxygen-radical-mediated injury
(98).
Iron, the third product of HO, is highly toxic through the Fenton reaction, which produces OH&*;
(99). Accordingly, there ought to exist some mechanism to stimulate the efflux from the cell of iron formed by HO. HO activity appears to be linked to cellular iron efflux. Thus, transfection of HO1 into mammalian cells stimulates iron efflux, and iron efflux is greatly diminished in fibroblasts from HO1 knockout mice
(84). These findings fit with evidence of low serum iron and accumulation of iron in the tissues of HO1 knockout mice
(100,
101). Whether HO2 similarly regulates iron efflux is not yet established, although HO2 knockout mice develop iron accumulation in their lungs in response to hyperoxia
(102).
Other gases might also function as neurotransmitters. Hydrogen sulfide (H
2S) exerts specific effects on the electrical properties of serotonin neurons in the dorsal raphe nucleus
(103). Abe and Kimura
(104) demonstrated that cystathionine beta-synthase produces H
2S in the brain. In addition, exogenous H
2S enhances NMDA neurotransmission and facilitates the induction of long-term potentiation in the hippocampus at concentrations comparable to those that occur physiologically
(104). Cystathionine beta-synthase is also expressed in smooth muscle and where physiologically relevant concentrations of H
2S enhance NO-mediated muscle relaxation
(105).
d-Amino Acids: Focus on d-Serine
Organic molecules, and therefore biological molecules, are based on the chemistry of the carbon atom. Carbon atoms can have up to four bonded groups attached to them in three-dimensional space, forming a tetrahedron. Because of this structure, carbon-containing molecules can have the same four constituents, yet differ in their structure by the location of the four groups in space. This property of carbon-based molecules is known as chirality. As an example, one’s hands are mirror images of each other, but they cannot be superimposed on each other. Similarly, carbon atoms with four different groups attached occur in two forms that are nonsuperimposable mirror images of each other, known as enantiomers. The elegant precision of biological reactions requires the recognition sites in proteins to be exceedingly specific. Just as a right-handed glove will not fit a left hand, enzymes and receptors can discriminate between the enantiomers of ligands and substrates.
As biology students, we were taught that organisms exclusively employ the
d-enantiomers for sugars, while amino acids for proteins are always in the
l-form.
d-Amino acids had been observed in bacteria and invertebrates
(106), but a role for
d-amino acids in higher species was deemed unlikely. However, observations in the 1990s have pointed to the existence of substantial quantities of some
d-amino acids in higher species, including humans
(107–
111). In the brain, levels of
d-serine are up to a third those of
l-serine, and, in a variety of tissues including the brain and certain glands,
d-aspartate levels are 20%–30% those of
l-aspartate
(107).
The existence of significant amounts of
d-serine and
d-aspartate in the brain suggested that some
d-amino acids serve specific neuroactive roles. Levels of
d-serine have marked variations in different regions of the brain, with highest concentrations in the forebrain, where NMDA-type glutamate receptors are enriched
(107). This finding meshed with certain known features of the NMDA receptor. For instance, Ascher and others had observed that the loss of NMDA receptor activation after rapid perfusion of neural preparations can be reversed by glycine
(112). Subsequent studies confirmed the existence of a glycine recognition site on the NMDA receptor and established that this receptor requires coactivation by glycine as well as glutamate
(113). These conclusions were somewhat puzzling, as glycine concentrations in the CNS are lowest in forebrain, areas that are enriched in NMDA receptors, and highest in the spinal cord and hindbrain, where glycine is known to function as an inhibitory neurotransmitter. We suspected that
d-serine might act as an endogenous ligand for the glycine site of the NMDA receptor, because numerous studies had established that
d-serine was at least as potent as glycine.
Based on these hints, we developed antisera to d-serine and mapped its localization in the CNS.
d-serine is indeed highly concentrated in areas of the brain enriched in NMDA receptors, where glycine levels are lowest
(114). In contrast, in some parts of the brain, such as the adult cerebellum and the accessory olfactory bulb, glycine, rather than
d-serine, may be associated with NMDA receptors
(115). Most striking was our observation that
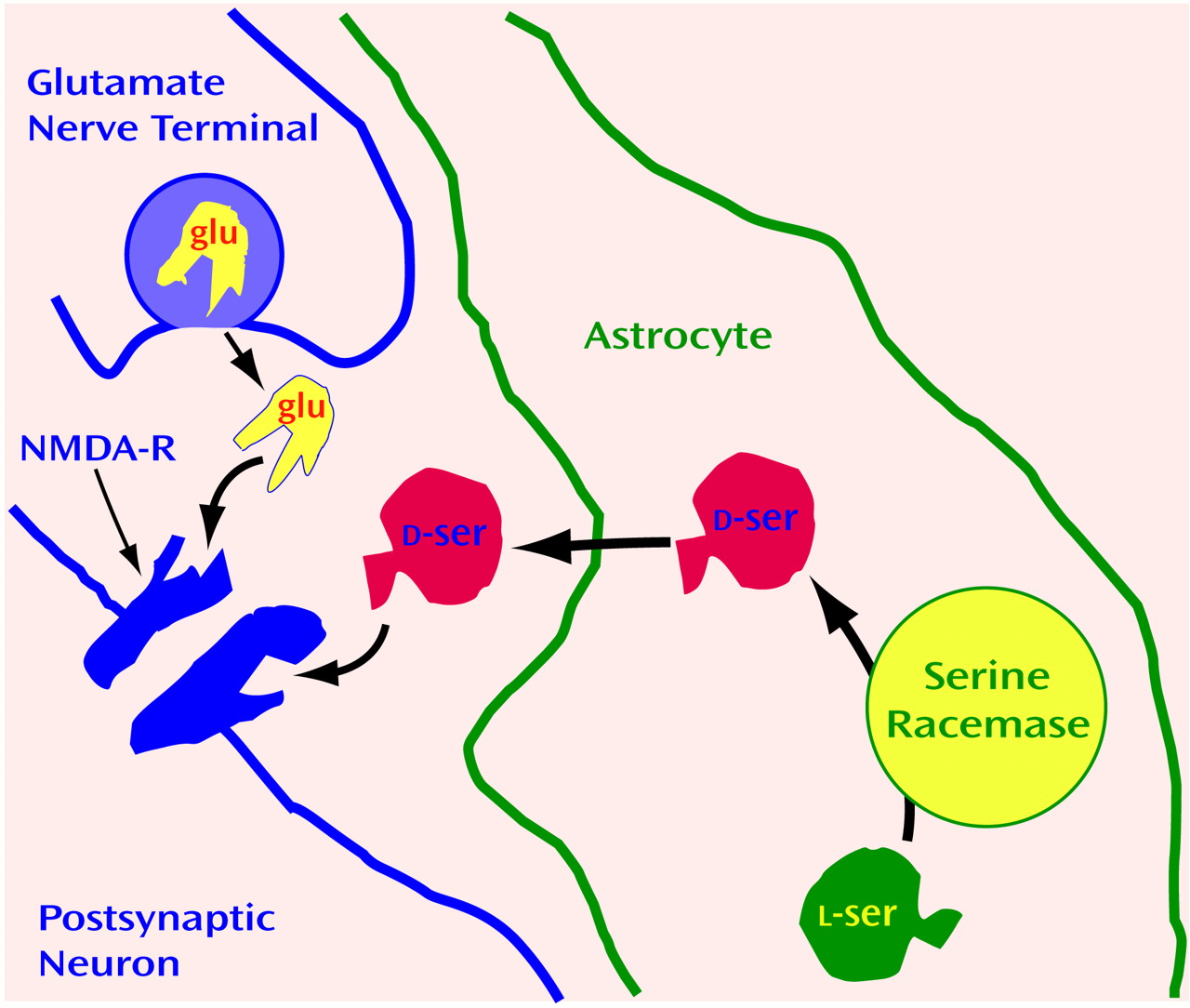
d-serine occurs in glia, not neurons.
d-Serine is exclusively localized to protoplasmic astrocytes that are enriched in gray matter together with NMDA receptors (
Figure 4). One reason that neurotransmitters have been presumed to derive exclusively from neurons is that only neurons were thought to release transmitters after appropriate excitation. In cultures of protoplasmic type II astrocytes, activation of non-NMDA glutamate receptors stimulates release of
d-serine
(114). In the brain, these astrocytes ensheathe the synapse so that any
d-serine released from astrocytes would have close proximity to NMDA receptors. Thus, we have proposed that synaptic release of glutamate from a presynaptic neuron triggers the release of d-serine from adjacent astrocytes to coactivate the NMDA receptors on nearby postsynaptic neurons (
Figure 4).
The idea that
d-serine possesses a physiologic role in the brain helped clarify a biochemical oddity first noticed in the 1930s. At that time, Hans Krebs
(118) discovered an enzyme that selectively deaminates
d-amino acids and designated it “
d-amino acid oxidase” (DAAOX). Because
d-amino acids were unknown in mammalian tissues, it was assumed that the enzyme was an evolutionary vestige from bacteria, that it was a mistake of nature, or that it served to oxidize glycine that is not chiral and therefore does not have enantiomeric forms. Our histochemical investigations showed marked regional variations in DAAOX localization, with concentrations exactly reciprocal to those of
d-serine
(114). These data suggest that DAAOX degrades
d-serine physiologically.
How might one definitively determine whether
d-serine is an endogenous modulator of NMDA receptor function? We utilized DAAOX as a tool, first establishing that
d-serine is the only substance in brain preparations degraded by DAAOX
(119). Then we showed that DAAOX treatment markedly reduces NMDA neurotransmission in cerebellar preparations by monitoring both NOS activity and cyclic GMP levels as well as by conducting electrophysiologic studies in slice and culture preparations of the cerebellum and the hippocampus
(119,
120)How might the brain synthesize
d-serine? After developing a specific assay to monitor
d-serine levels, we isolated an enzyme from mammalian brain that converts
l-serine to
d-serine, designated it serine racemase
(116), and then cloned its complementary DNA
(117). Serine racemase is localized to the
d-serine-containing protoplasmic astrocytes in areas of the brain enriched in NMDA receptors
(117). It is a novel protein, although it possesses some modest sequence similarity to other enzymes that use serine as a substrate. These enzymes, like serine racemase, require pyridoxal phosphate (vitamin B
6) as a cofactor
(116,
117).
d-Serine challenges various dogma about neurotransmitters, perhaps to a greater extent even than NO and CO. The idea of a function for
d-amino acids in mammals, especially as a neurotransmitter, goes against long-standing biochemical and physiological teachings. Does
d-serine satisfy criteria for a neurotransmitter, shown in
Appendix 1? It is localized to the sites of its receptors and possesses a dedicated biosynthetic enzyme.
d-Serine is released on appropriate stimulation and mimics the actions of the physiologic transmitter. Thus, in many ways,
d-serine fulfills more criteria than many neuropeptides that are well accepted as transmitters. However, the concept of a neurotransmitter arising from glia may discomfort some neuroscientists. Glia do not have synaptic vesicles, so
d-serine release cannot occur through classic exocytosis. Instead, its release may result from reversing the directionality of an amino acid transporter that might otherwise remove substances from the synapse. Thus, as for NO and CO, aberrant release processes would confound those who insist that neurotransmitters adhere to traditional criteria.
The cloning of serine racemase provides a potentially powerful approach to learning about d-serine in the brain and to exploring therapeutic ramifications. Future studies will allow monitoring of NMDA neurotransmission, long-term potentiation, and overall behavior in serine racemase gene knockout mice. Inhibitors of serine racemase would be expected to diminish NMDA neurotransmission, and so, like NMDA receptor antagonists, serine racemase inhibitors might be beneficial in treating stroke and other conditions associated with excess excitation. Glutamate neurotoxicity may be relevant for the therapy of neurodegenerative diseases, whether or not excitotoxicity is directly involved in pathophysiology. We don’t know the precise etiology of conditions such as Parkinson’s disease and Alzheimer’s disease. However, regardless of the initiating event, neurons do degenerate in patients with these disorders. Because glutamate release is augmented in the presence of hypoxic neural damage, it is likely that excessive levels of glutamate are released as the disease progresses and that glutamate may work together with other insults to permit cell death. Concievably, diminishing glutamate receptor activation may block the progression of neurodegenerative conditions such as Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, and amyotrophic lateral sclerosis (ALS). Although reliable animal models of Alzheimer’s disease and Huntington’s disease have been difficult to establish, there are model systems for ALS and Parkinson’s disease. Glutamate receptor antagonists are therapeutic in these models, and thus serine racemase inhibitors merit examination as well.
d-Serine and NMDA transmission may be relevant to schizophrenia. The psychotic state after administration of NMDA antagonists such as phencyclidine (PCP) closely resembles certain features of schizophrenia, more than most drug psychoses. Whereas psychoses elicited by drugs such as amphetamines and cocaine manifest positive schizophrenic symptoms, psychoses associated with PCP elicit negative as well as positive symptoms
(121). Besides causing the negative syndrome of emotional withdrawal, PCP elicits cognitive dysfunction characteristic of schizophrenia. Evidence for a relationship between glutamatergic and dopamine models of schizophrenia comes from studies showing altered dopamine release after PCP administration
(122,
123).
According to the NMDA receptor model of schizophrenia, one would expect glutamate agonists to be therapeutic. Since glutamate itself is potentially toxic and too rapidly metabolized, researchers have administered glycine,
d-serine, or cycloserine (which mimics
d-serine at NMDA receptors) to assess their potential efficacy in patients with schizophrenia. Beneficial effects have been reported
(124,
125).
d-Serine may not be the only
d-amino acid neurotransmitter candidate.
d-Aspartate was first discovered in invertebrates
(126,
127). Several groups identified
d-aspartate in mammalian tissues with notable concentrations in endocrine glands and the brain
(128–
131). D’Aniello and associates
(132,
133) observed that
d-aspartate is released from the testes and may stimulate testosterone synthesis, and some studies have found changes in
d-aspartate levels in the brains of patients with Alzheimer’s disease
(134,
135). With antisera to
d-aspartate, we localized the amino acid to selected neuronal populations in the brain as well as to epinephrine-containing cells in the adrenal medulla, to the vasopressin-releasing hypothalamic neurons innervating the posterior pituitary gland, and to pinealocytes in the pineal gland
(136). Although
d-aspartate can activate NMDA receptors, the localizations of
d-aspartate do not match those of NMDA receptors, and
d-aspartate levels are far lower than glutamate levels. Thus, at present, too little is known about
d-aspartate to draw any firm conclusions about its function in endocrine glands or the brain.
Conclusions
This essay has focused on NO, CO and d-serine as recently identified candidate neurotransmitters. A historical review reveals that every neurotransmitter candidate after acetylcholine has altered one or more preconceptions about the defining characteristics of a neurotransmitter. With so much controversy, one may be tempted to discard the term. Alternatively, bearing in mind these historical lessons, we might try adopting a reasonably liberal conceptualization of a neurotransmitter, such as the following: A transmitter is a molecule, released by neurons or glia, that physiologically influences the eletrochemical state of adjacent cells. Outside the CNS, those adjacent target cells need not be neurons and, in most instances, would be smooth muscle or glandular cells. In considering the CNS, researchers usually think of neurotransmitters as influencing adjacent neurons, but one need not exclude influences on glia or blood vessels. Advocating changes in classic definitions of neurotransmitters may seem heretical. However, previous assumptions about neurotransmission have been challenged repeatedly over the past 50 years. In all of these instances, we have benefited through new insights with important therapeutic implications for neuropsychiatry.