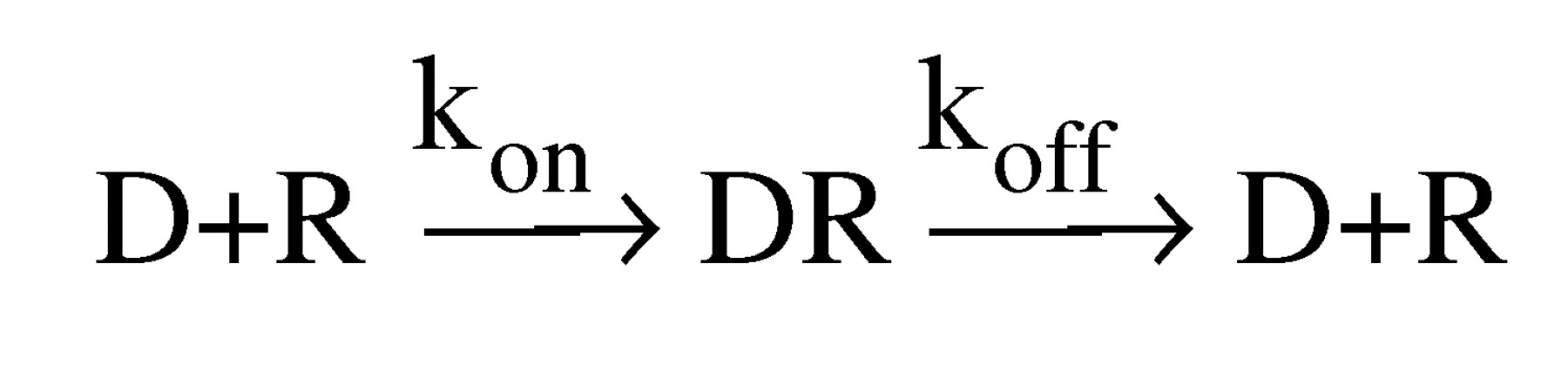This Orwellian phrase captures the essence of our hypothesis. We propose that a faster dissociation from the D2 receptor, without reference to any other receptor system, explains the most pertinent aspect of what is currently called “atypical” antipsychotic activity. We present our thoughts in five sections. In the first, we define “atypicality” and briefly review some technical concepts regarding receptor pharmacology relevant to our discussion. Next, we review positron emission tomography (PET) studies in patients and derive from them the centrality of D2 occupancy in generating antipsychotic response. The third section reviews data showing how previous hypotheses regarding the role of serotonin 5-HT2 and dopamine D4 were actually driven by underlying low affinity and fast dissociation from the D2 receptor. In the fourth and fifth sections we explain how fast dissociation of a drug from the D2 receptor, at molecular and system levels, leads to the atypical antipsychotic effect. Finally, we point out the limitations of our current hypothesis, how it relates to the role of other receptors, and the implications of this hypothesis for future drug development.
The Molecular Basis of Atypicality
To our knowledge, all efforts to produce antipsychotic action without D
2 blockade have been unsuccessful. Notable recent failures include drugs that act only on 5-HT
2 (MDL-100907)
(37), D
4 (L-745,850)
(42), 5-HT
2 and D
4 (fananserin)
(36), and dopamine D
1 receptors (SCH-23390)
(43). Thus, currently, D
2 blockade remains a necessary and sufficient condition for antipsychotic action. Since all antipsychotics block D
2 receptors, it has often been thought that atypical antipsychotics must differ by involving a separate receptor mechanism. Two hypotheses, one invoking the role of the 5-HT
2 receptor and another proposing a role for the D
4 receptor, have been particularly influential.
In an important article that supported the 5-HT
2/D
2 hypothesis, Meltzer et al.
(44) studied the in vitro binding profile of 37 antipsychotics (some were clinically proven; others were preclinical at that time). This report constitutes the cornerstone of the serotonin-dopamine hypothesis of atypicality. It showed that atypicals had a greater difference between their 5-HT
2 and D
2 affinities than did typicals. However, what is often overlooked is that the greater difference between 5-HT
2 and D
2 affinities in atypicals was due not to higher 5-HT
2 affinity but to lower D
2 affinity. For the 20 typicals and 17 atypicals in that report, the mean affinity at the 5-HT
2 receptors (expressed as pKi) was 8.37 (SD=0.16) (for typicals) and 8.36 (SD=0.25) (for atypicals); there was no statistical difference between the two. On the other hand, their D
2 mean affinities were 8.88 (SD=0.14) (for typicals) and 7.02 (SD=0.25) (for atypicals); the difference was highly significant (F=43.5, df=1, 35, p<0.001). Thus, atypical antipsychotics were shown to differ from typicals by their lower affinity at the D
2 receptor, not by their higher affinity at 5-HT
2. The same pattern of results was found in in vivo measures of affinity in animal models
(41). A discriminant function analysis in the article showed that a low D
2 affinity was by far the single biggest contributor to atypicality and that the contribution of high 5-HT
2 affinity was clearly secondary
(44).
For the sake of argument, one could suggest that even though low D
2 affinity drives atypicality, the presence of high 5-HT
2 affinity is still necessary (the 5-HT
2/D
2 ratio argument
[44]). Several lines of evidence refute this possibility. First, atypical antipsychotics such as amisulpride (available in Europe)
(34,
45,
46) and remoxipride (withdrawn from clinical use because of association with aplastic anemia)
(47) provide atypical clinical benefits with no relevant affinity for the 5-HT
2 receptor. Amisulpride is particularly interesting since it has been shown to be as effective as haloperidol in the treatment of positive symptoms; it has fewer extrapyramidal side effects
(46) and more effectiveness in treating negative symptoms
(48,
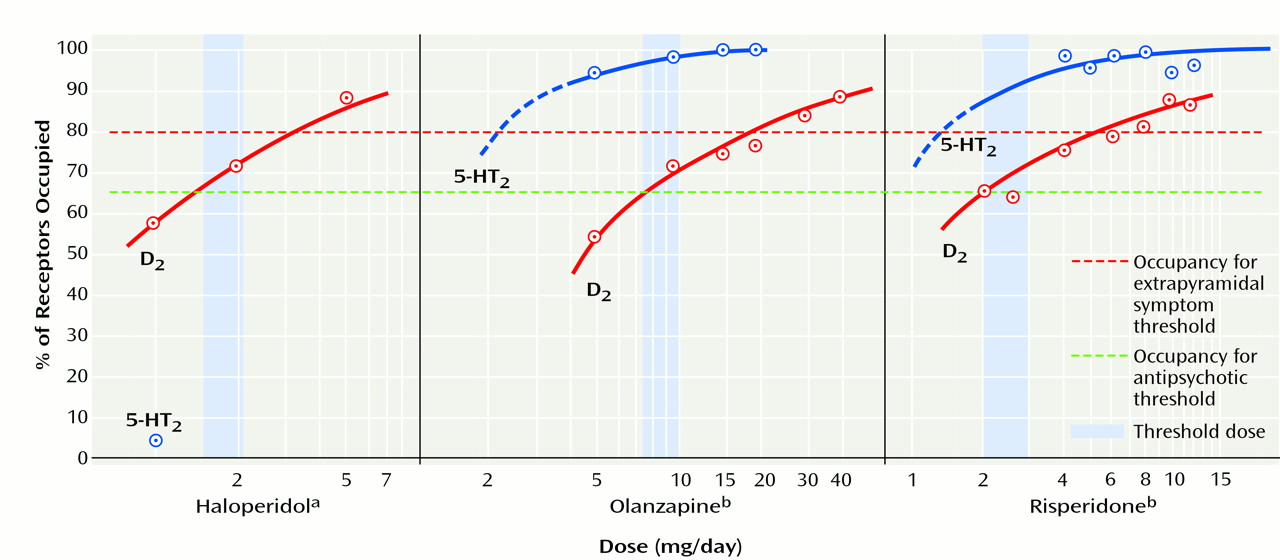
49), thus matching the clinical profile of multireceptor atypicals such as risperidone and olanzapine but without any effects at other receptors. Second, the 5-HT
2/D
2 ratio argument is not compatible with human PET data. Subtherapeutic doses of olanzapine (<5 mg/day) and risperidone (<2 mg/day) have high 5-HT
2 versus D
2 occupancy ratios; however, these doses are not even antipsychotic, let alone atypical. Thus, high 5-HT
2 occupancy seems neither necessary nor sufficient for the atypical antipsychotic effect.
Antagonism at the D
4 receptor has also been mentioned as a basis for atypicality. However, when this hypothesis was considered in the context of other typical and atypical drugs, it also suffered from same problems as the 5-HT
2/D
2-ratio hypothesis. The affinity of typical drugs such as haloperidol and chlorpromazine for the D
4 receptor is actually higher than that of drugs such as clozapine or olanzapine
(50). Thus, typical antipsychotics are more potent than atypicals, not less potent, at the D
4 receptor. In light of this, a D
4 basis of atypicality cannot be sustained unless one links it to D
2 and then argues for a high D
4/D
2 ratio
(51). The D
4/D
2 ratio argument (like the argument for the 5-HT
2/D
2 ratio) also falls short of evidence. First, the differences in the ratio are driven by differences in D
2, not by differences in D
4, since haloperidol is more potent than clozapine at the D
4 receptor
(50). Second, drugs with practically no affinity for the D
4 receptor, for example, quetiapine and amisulpride
(50–
52), are atypical antipsychotics. Finally, clinical trials of drugs selective for the D
4 receptor
(42) or those that combine 5-HT
2 and D
4 selectivity
(36) do not show evidence of antipsychotic efficacy. Thus, high D
4 affinity is neither necessary nor sufficient for producing atypical antipsychotic activity.
We propose that a low affinity at the D2 receptor in and of itself, without reference to any other receptor profile, is sufficient for producing atypical antipsychotic activity. Previous hypotheses regarding a role for the 5-HT2/D2 and D4/D2 ratios seemed valid because they shared a common denominator: a low affinity for D2. The current generation of atypical antipsychotics are atypical not because they have high levels of 5-HT2 or D4 activity but because they have a low affinity at the D2 receptor. That raises the question, why should a low affinity at the D2 receptor give rise to atypicality?
Affinity (more precisely, K
d) is, by definition, the ratio of k
off/k
on (the rate at which the drug moves off of and on to the receptor). In theory, either a difference in k
on or a difference in k
off could lead to low affinity. To examine whether k
on or k
off drives the differences in D
2 affinity between typical and atypical antipsychotics; we measured the affinity, k
on and k
off, for a series of typical and atypical antipsychotics
(53). Although affinity for the D
2 receptor varied nearly a thousand-fold, from 0.025 nM for nemonapride to 155 nM for quetiapine, 99% of the difference in affinity of the antipsychotics was driven by differences in their k
off at the D
2 receptor. Differences in k
on did not account for any significant differences in affinity
(53). All antipsychotics (typical or atypical) attach to the D
2 receptor with a similar rate constant; they differ only in how fast they come off of the receptor. We propose that this relationship between fast k
off and low affinity is the critical underlying molecular feature that explains why low affinity at the D
2 receptor leads to the atypical antipsychotic effect.
In test tube experiments, dissociation of an antipsychotic from the receptor is determined mainly by the molecular properties of the drug. In the living brain, this molecular property of fast dissociation displays itself on a stage of fluctuating plasma/brain levels of the drug. For example, clozapine, even at steady-state levels, shows a peak of 850 ng/ml and a trough of 300 ng/ml daily. Thus, in a living system the dissociation of a drug from a receptor is determined by phenomena at two levels: events at a molecular level in the synapse (determined mainly by koff) and events at a system level (which is a complex function of plasma half-life, brain half-life, and koff). We will distinguish between these two levels of fast dissociation (one molecular and one systemic) and point out the relevance of each to atypicality.
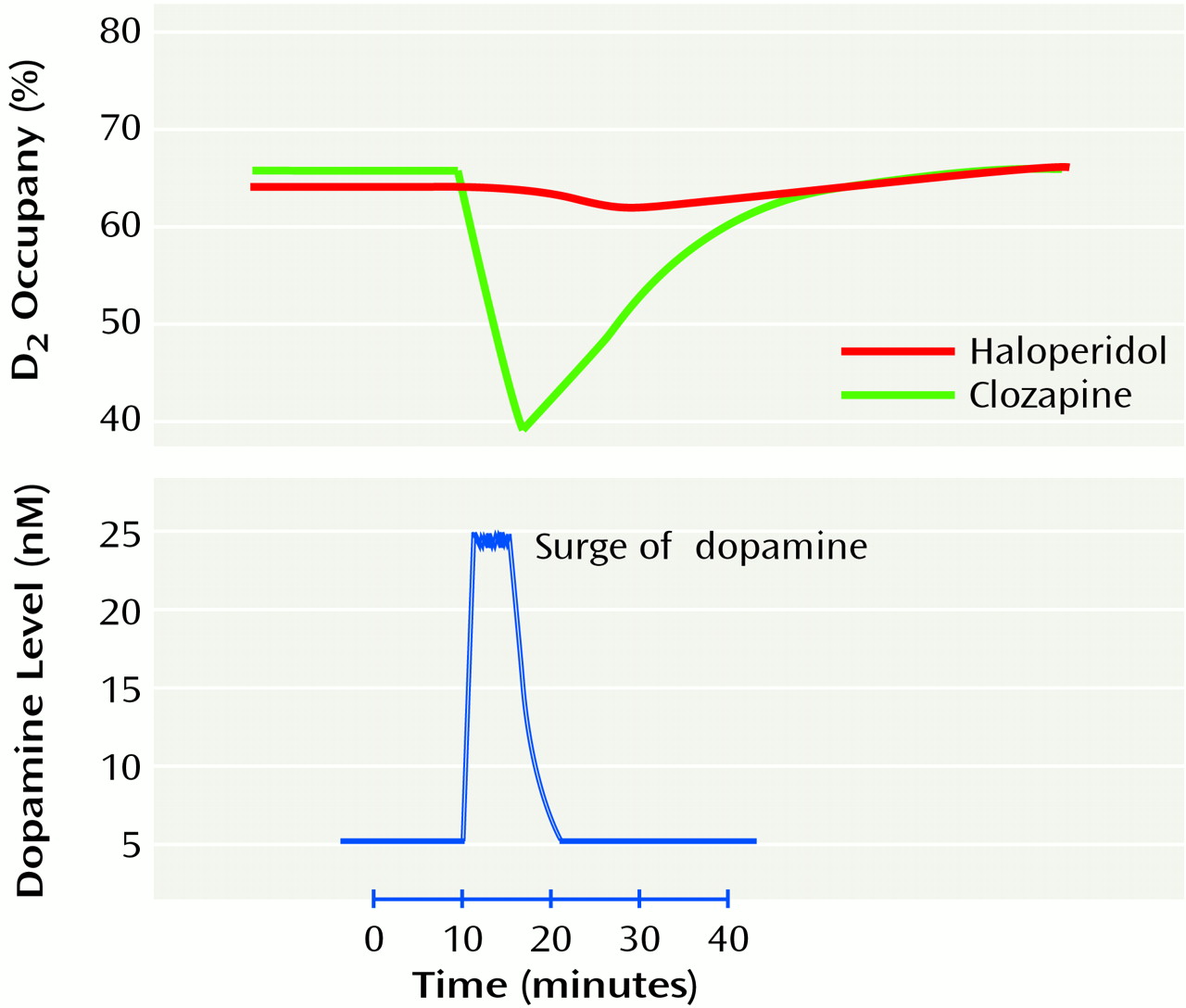
Clozapine, the quintessential atypical antipsychotic, shows clear evidence of a definite, yet transient, effect by means of the D
2 system. Clozapine causes a clear dose-dependent elevation of dopamine turnover and release, indicating relevant dopamine blockade
(62,
63). However, the effects of atypicals are much more transient than those of typical antipsychotics. In studies examining brain occupancy it has been shown that clozapine shows a rapid and transient D
2 occupancy
(62,
63), whereas haloperidol shows prolonged occupancy in the same models. As an example, Saller and Salama
(63) found that 40 mg/kg of intraperitoneal clozapine produced 61% D
2 occupancy within 30 minutes; this decreased to 0% in 4 hours. Haloperidol (1 mg/kg intraperitoneal) produced 57% occupancy at 30 minutes and was still at 62% at 4 hours. Similar and parallel evidence of transience is found with prolactin elevation. For example, both clozapine (20 mg/kg intraperitoneal) and haloperidol (0.25 mg/kg intraperitoneal) led to robust prolactin elevation in rats (from a baseline of <10 ng/ml to a peak of 70 ng/ml for both clozapine and haloperidol at 30 minutes
[64]). However, the clozapine-induced prolactin elevation was completely back to normal at 2 hours, whereas the haloperidol level was still elevated (approximately 70 ng/ml) at 4 hours after receipt of the drug
(64).
More recently, we replicated this pattern of findings in patients and produced significant transient prolactin elevations with the atypical antipsychotics clozapine, quetiapine, and olanzapine
(23,
65). We found that even with prolactin-sparing atypicals such as olanzapine, clozapine, and quetiapine, almost all patients showed prolactin elevation (some of them in the abnormal range) 2–6 hours after receiving the last dose of drug. However, the prolactin levels declined rapidly and returned to normal by 12–24 hours after receipt of the last dose
(23,
65). Thus, it is not that atypical antipsychotics do not cause prolactin elevation; they just cause a transient prolactin elevation, indicative of the transient functional effects of D
2 blockade. Since the prolactin elevation subsides by the time of the next dose, it does not lead to drug accumulation and sustained high levels, as has been observed with prolactin-raising antipsychotics.
In short time frames, i.e., seconds to minutes, dissociation is largely determined by the k
off of a drug. However, over longer time frames, i.e., hours and days, drug levels in the brain also change. Thus, in addition to fast dissociation at a molecular level, drugs also show different patterns of system-level occupancy, depending largely on the manner in which their brain levels change with drug administration. Even for a given drug (i.e., with molecular k
off fixed), different temporal patterns of system-level occupancy have important differential consequences. Kashihara et al.
(66) administered a given dose of haloperidol by means of a daily injection (which gives rise to transiently high system-level occupancy) or with a subcutaneous pump (which provides sustained system-level occupancy) and found that the sustained mode of administration led to significantly higher D
2 receptor up-regulation and induced tolerance to subsequent dopamine blockade. See and Ellison
(67) found that sustained administration led to the development of tardive dyskinesia-like motor symptoms (suggestive of up-regulation), whereas intermittent (weekly) administration did not produce these motor symptoms and, if anything, led to dystonia-like acute extrapyramidal symptoms suggestive of greater sensitivity to dopamine blockade. In several other studies
(66–
69) a similar pattern was observed: sustained blockade led to tolerance and up-regulation, whereas transient occupancy avoided tolerance and up-regulation and made the system more sensitive to the antidopaminergic effects of antipsychotics. This differential response to transient versus sustained occupancy probably reflects an underlying property of the dopamine system, since a similar pattern of changes has also been observed with dopaminergic agonists in the context of Parkinson’s disease or drug abuse
(70–
72).