In patients with posttraumatic stress disorder (PTSD), low cortisol levels have repeatedly been reported despite high central nervous system corticotropin-releasing hormone (CRH) activity
(1–
3). An enhancement of glucocorticoid-receptor-mediated negative feedback in the pituitary of PTSD patients, as demonstrated with the dexamethasone suppression test
(4), might explain this phenomenon. However, hippocampal mineralocorticoid receptors, which have a higher affinity for cortisol than do glucocorticoid receptors, exert an inhibitory influence on hypothalamic-pituitary-adrenal (HPA) axis activity
(5,
6). Recently, the possibility of dynamic regulation of mineralocorticoid receptors was demonstrated in a preclinical study showing an increase in hippocampal mineralocorticoid receptor density after psychological stress
(7). However, so far no investigation has characterized the role of mineralocorticoid receptors in PTSD. Based on our hypothesis that PTSD patients exhibit an elevated mineralocorticoid receptor function, we predicted an amplified cortisol response in PTSD patients compared to healthy subjects after administration of the mineralocorticoid receptor antagonist spironolactone, and enhanced cortisol secretion after subsequent stimulation with CRH.
Method
Subjects were 12 outpatients with PTSD (seven women, five men, mean age=37.2 years, range=24–52) who had no other primary anxiety or primary mood disorder and no psychotic, organic, or substance-related disorders, as assessed with the Structured Clinical Interview for DSM-IV, and 12 healthy sex- and age-matched comparison subjects (mean age=36.3 years, range=22–54). The subjects had been free of medication for at least 14 days and had undergone a thorough medical examination to rule out other illnesses, drug intake, or lifestyle factors that could interfere with the study. The comparison subjects had no personal or family history of any major psychiatric disorder. The protocol was approved by the local institutional review board, and each participant provided written informed consent before the investigation.
In a single-blind, fixed-order design, subjects were pretreated orally with placebo and 200 mg of spironolactone (Hoffmann-La Roche, Grenzach-Wyhlen, Germany) at 9:00 a.m. and noon on the first and second days of the study, respectively. On both study days an intravenous catheter was placed in the subject’s arm at 1:00 p.m., and the subject remained supine in a soundproof room at the hospital until 5:00 p.m. A bolus injection of 100 μg of human CRH (Clinalfa, Läufelfingen, Switzerland) was given at 3:00 p.m. Blood samples were drawn at 2:30, 2:45, 3:00, 3:15, 3:30, 3:45, 4:00, 4:30, and 5:00 p.m. to determine cortisol and adrenocorticotropic hormone (ACTH) levels. Samples were placed on ice, plasma was separated immediately, and specimens were stored at –80°C until analysis. Blood pressure was registered at blood sampling times with an automatic device.
Plasma cortisol levels were determined by using a radioimmunoassay (ICN Biomedicals, Carson, Calif.). ACTH concentrations were determined with an immunoradiometric assay (Nichols Institute, San Juan Capistrano, Calif.). Intra- and interassay coefficients of variation were less than 8% for both assays. Cross-reactions of spironolactone and the spironolactone metabolite canrenoate at concentrations up to 4.0×105 ng/ml were below 0.2% in the cortisol assay.
For statistical comparisons by multivariate analyses of variance (MANOVA), the following variables were calculated from the endocrine and blood pressure data: the mean basal levels from 2:30 to 3:00 p.m. and the maximal change in level and the area under the curve minus linear background between 3:00 and 5:00 p.m., i.e., after CRH stimulation. Data are given as means and standard deviations. The factors in the MANOVA were treatment (spironolactone versus placebo, as the within-subjects factor) and group (PTSD versus comparison subjects, as the between-subjects factor). To avoid colinearities, the parameters of cortisol level, ACTH level, and systolic and diastolic blood pressure were tested in separate MANOVAs. When a significant main or interaction effect was found, a univariate F test was used to identify the variables that contributed significantly to the effect. An alpha of 0.05 was accepted as the level of significance. All post hoc tests were performed at a reduced level of significance according to the Bonferroni procedure.
Results
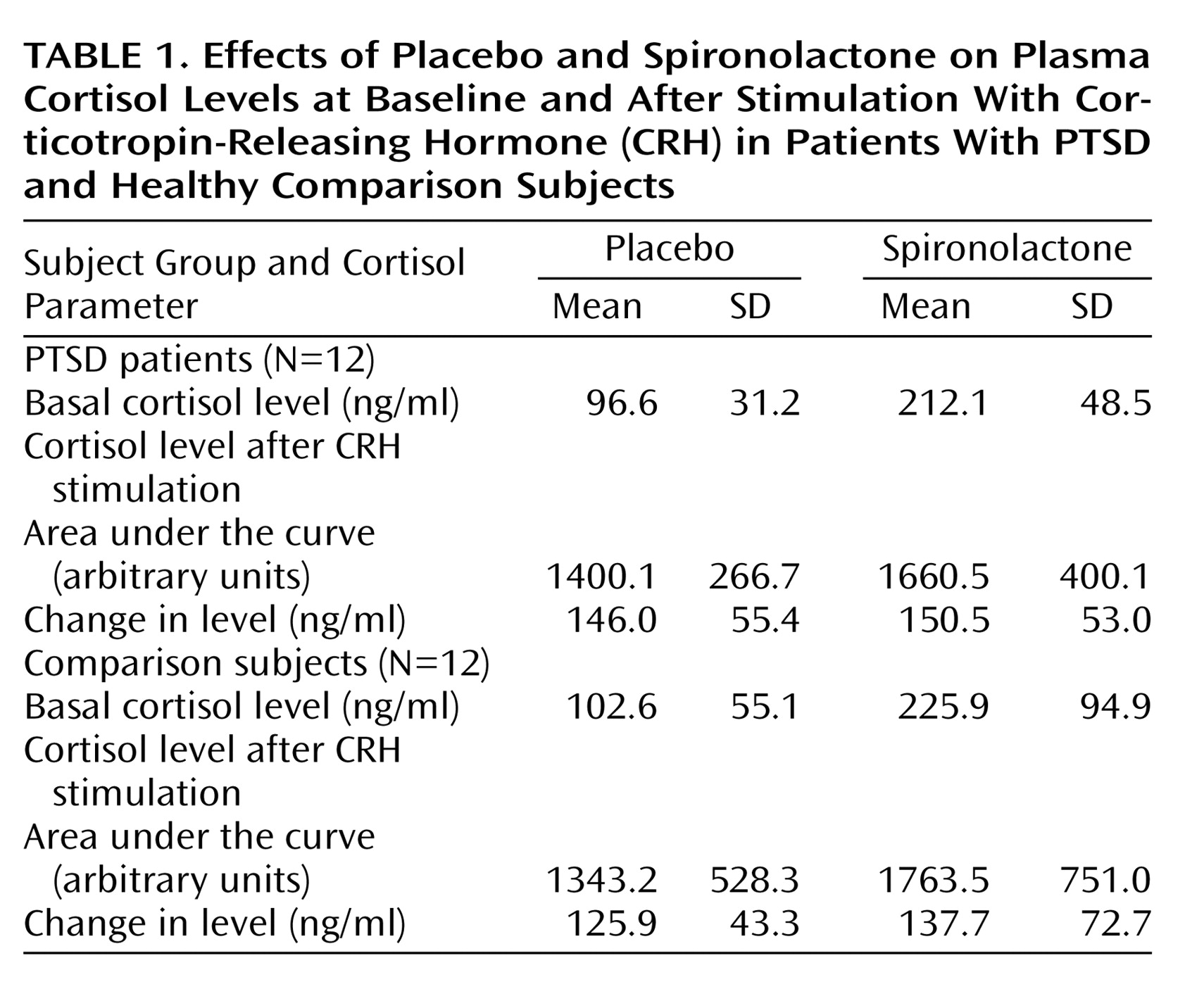
Cortisol parameters revealed significant treatment effects of spironolactone, compared with placebo (F=22.3, df=3, 20, p=0.001), for mean basal cortisol level and the area under the curve (F=43.2 and F=35.1, respectively, df=1, 22, p=0.001 each) but not for the change in level (F=0.6, df=1, 22, p=0.47). However, neither a significant group effect (F=0.4, df=3, 20, p=0.76) nor a significant group-by-treatment effect (F=0.7, df=3, 20, p=0.54) was detected (
Table 1).
For ACTH parameters a significant treatment effect emerged (F=83.8, df=1, 22, p=0.001) for mean basal level (F=83.8, df=1, 22, p=0.001) but not for the area under the curve (F=0.0, df=1, 22, p=0.93) or the change in level (F=2.2, df=1, 22, p=0.15). The mean basal ACTH levels in the placebo versus spironolactone condition were 16.6 pg/ml (SD=9.7) versus 33.0 pg/ml (SD=11.8) in PTSD patients and 15.9 pg/ml (SD=8.0) versus 31.1 pg/ml (SD=10.4) in comparison subjects. No group effect (F=0.9, df=3, 20, p=0.51) or group-by-treatment effect (F=0.9, df=3, 20, p=0.78) was found.
The spironolactone action on cortisol and on ACTH was not caused by blood pressure effects, since systolic and diastolic blood pressure showed no treatment effect (F=0.9, df=3, 20, p=0.62 and F=0.9, df=3, 20, p=0.46, respectively) or group-by-treatment effect (F=0.9, df=3, 20, p=0.40 and F=0.9, df=3,20, p=0.50, respectively).
Discussion
Our findings of increased spontaneous and CRH-stimulated plasma cortisol after acute treatment with the mineralocorticoid receptor antagonist spironolactone are similar to those of previous studies in humans
(8,
9). However, no differential effects of spironolactone in PTSD patients versus healthy comparison subjects were detected. Due to the limitations in our study design, we were unable to determine whether the lack of group differences was related to a lack of mineralocorticoid receptor up-regulation in PTSD or to factors such as compensatory neuropeptide regulation or hyperactive pituitary glucocorticoid receptor-mediated negative feedback
(4) masking the putative mineralocorticoid receptor changes in our patient group. Also, in contrast to expectation, postplacebo basal cortisol levels in the patients were indistinguishable from those in the normal comparison subjects. Spironolactone administration closer to the circadian nadir of cortisol levels, when HPA axis tone is primarily driven by mineralocorticoid receptors, might better distinguish the groups.
Further investigation of mineralocorticoid receptor function in PTSD may be indicated, since mineralocorticoid receptors modulate pertinent cognitive and behavioral effects
(5). In particular, given an anxiolytic action of hippocampal mineralocorticoid receptor blockade in rodents
(10), mineralocorticoid receptor function might be a potential target for psychopharmacological intervention.