Hypotheses invoking the role of dopamine in Tourette’s syndrome have suggested abnormalities of both post- and presynaptic function, such as supersensitive dopamine (D) receptors, dopamine hyperinnervation, and abnormal presynaptic terminal function. The possibility of abnormal postsynaptic receptors was initially suggested by findings of reduced basal and turnover levels of a dopamine metabolite, homovanillic acid, in cerebrospinal fluid that were restored to normal after the administration of haloperidol
(6,
8,
9). Limited studies of D
1 and D
2 receptor binding in postmortem striatal tissue have shown differences between Tourette’s syndrome and comparison subjects, but none that reached significance
(7). Investigations of D
2 receptors by PET and single photon emission computed tomography (SPECT) have produced inconsistent findings in studies involving Tourette’s syndrome patients and comparison subjects
(10–
13). Two studies have supported the hypothesis that the dopamine receptor is involved in the neurobiology of Tourette’s syndrome. In a study of five sets of identical twins, greater [
123I]iodobenzamide ([
123I]-IBZM) binding observed in the head of the caudate nucleus was associated with greater tic severity
(12). In our PET study with a spiperone derivative, [
11C]3-N-methylspiperone, B
max levels above the 95th percentile prediction limit (normal regressed against age) were observed in four of 20 adult subjects, and multiple linear regression analyses revealed a trend between the severity of vocal tics and B
max values
(13). In contrast to studies reporting D
2 receptor changes, other studies in which [
123I]-IBZM or [
11C]raclopride was used did not show differences
(10,
11).
Attempts to provide support for postulated dopamine hyperinnervation (i.e., greater number of dopamine terminals) by PET or SPECT binding have resulted in conflicting reports. For example, a small study that used [
123I]β-CIT SPECT found that striatal dopamine transporter binding was higher in five adults with Tourette’s syndrome than in a group of comparison subjects
(14). In a second study that used a similar technique, the mean striatal activity ratio was significantly higher in Tourette’s syndrome patients, and five of 12 Tourette’s syndrome subjects showed striatum/occipital cortex ratios more than two standard deviations above the normal mean
(15). In contrast, other investigators who used SPECT or PET techniques in adult patients with Tourette’s syndrome reported no difference in dopamine transporter binding relative to that seen in comparison subjects
(16,
17). Studies evaluating dorsal striatal dopaminergic innervation, by use of in vivo measures of vesicular monoamine transporter type 2 binding with the ligand (+)-alpha-[
11C]dihydrotetrabenazine, showed no differences between Tourette’s syndrome subjects and age-compatible normal comparison subjects
(18). These results suggest that there is no increase in striatal innervation but do not exclude an abnormality in the regulation of dopamine release or reuptake. Last, a third dopamine proposal implicates a presynaptic dopamine abnormality involving dopa decarboxylase activity. In a PET study, 11 adolescents with Tourette’s syndrome accumulated [
18F]fluorodopa at a level 25% higher in the left caudate nucleus and 53% higher in the right midbrain relative to levels seen in comparison subjects
(19). The authors suggested that upregulation of dopa decarboxylase activity could explain these alterations and that the process reflects deficits in a variety of functional elements of the dopamine system.
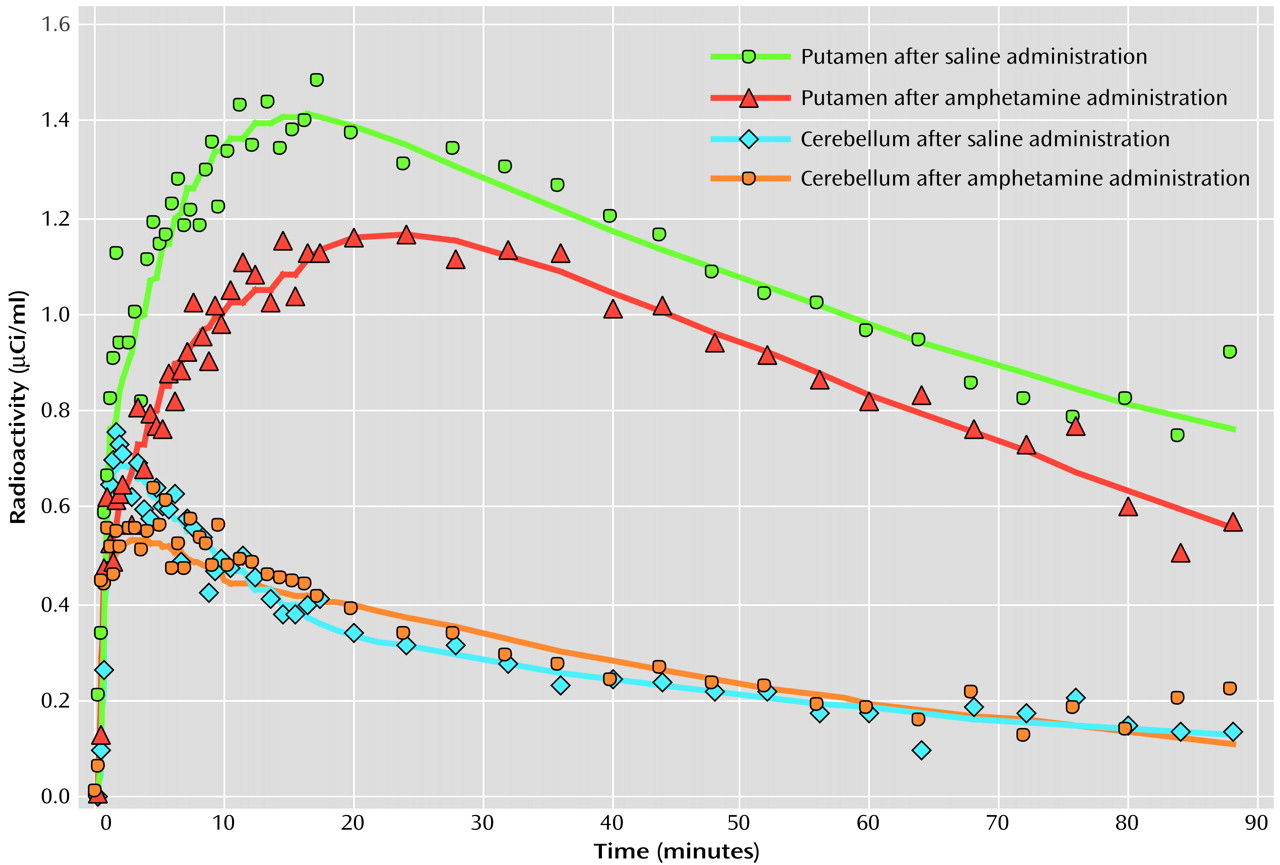
In order to evaluate dopamine release in Tourette’s syndrome, we intravenously administered a central stimulant (amphetamine) that enhances dopamine release and blocks its reuptake
(20). Analogous studies with such a provocative stimulant challenge have been performed in rodents
(21), baboons
(22), normal humans
(23), and in patients with schizophrenia
(24,
25) and cocaine dependence
(26). In the current study, each subject underwent two [
11C]raclopride PET scans: one after intravenous administration of saline and the second after amphetamine was intravenously administered to induce dopamine release. Studies that have used a stimulant challenge followed by either PET or SPECT binding have demonstrated greater dopamine release in individuals with schizophrenia
(24,
25) and decreased release in those with cocaine dependence
(27). Application of the amphetamine stimulation technique to adults with Tourette’s syndrome suggests that tics may be associated with higher intrasynaptic levels of dopamine.
Discussion
Dopamine has an important influence on frontal-subcortical neurotransmission, either through presynaptic effects on corticostriatal afferents or postsynaptic effects on striatal neurons. In Tourette’s syndrome, dopamine hypotheses have been proposed that suggest either an excess of dopamine or an increased sensitivity to the neurotransmitter. Although several SPECT and PET studies have suggested that some individuals may have supersensitive D
2 receptors
(12,
13), concerns about these investigations include the effect of prior pharmacotherapy and the lack of compelling evidence that this is the primary abnormality in the majority of subjects. In the present investigation, similar to a prior PET study with [
11C]raclopride
(11) and a SPECT study with [
123I]-IBZM
(10), mean binding potential following the saline infusion did not significantly differ between Tourette’s syndrome and comparison subjects. These results could be interpreted as reflecting normal D
2 receptor levels; however, several investigators have claimed that benzamide radioligands have greater susceptibility to endogenous dopamine that may, in turn, mask B
max differences
(34,
35). A second hypothesis, which proposes dopaminergic hyperinnervation, is negated in part by findings of normal presynaptic striatal dopamine terminals as determined by quantifying regional densities of dopamine transporter and vesicular monoamine transporter type 2
(17,
18). Nevertheless, although there may be normal numbers of dopamine terminals, a functional alteration of dopamine reuptake or dopamine release remains possible.
Volkow and colleagues
(23,
27) pioneered the use of a methylphenidate plus [
11C]raclopride PET binding method to measure intrasynaptic dopamine content and subsequently showed levels to be diminished in cocaine users. Laruelle et al.
(24) substituted intravenous amphetamine for methylphenidate and, using [
123I]-IBZM SPECT, reported significantly greater dopamine release in adult schizophrenic patients than in normal comparison subjects. The fixed scan order (saline first, then amphetamine) is the standard protocol for all such bolus studies and is not considered a potential confound
(23,
36,
37). In fact, the initial use of amphetamine could result in a possible carryover effect and a necessary prolonged delay before the saline study. A smaller magnitude of change in our comparison subjects, relative to that reported in other protocols
(24,
25,
38), is believed to be due to our use of a bolus injection of amphetamine that was not followed by a constant infusion.
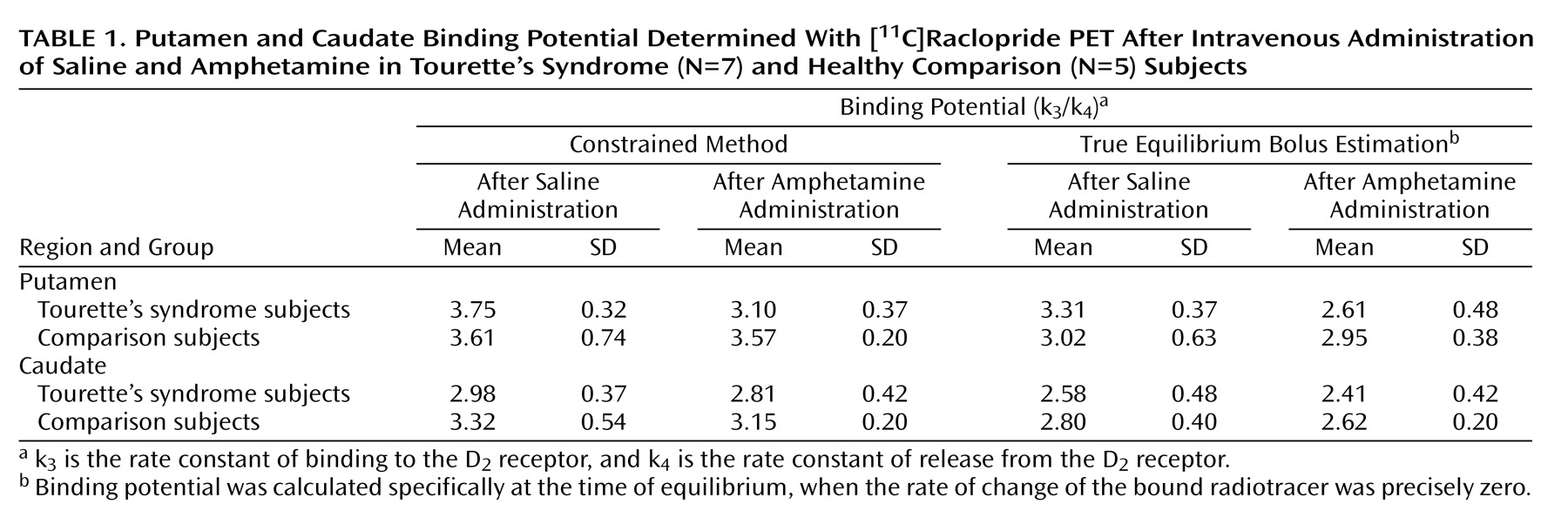
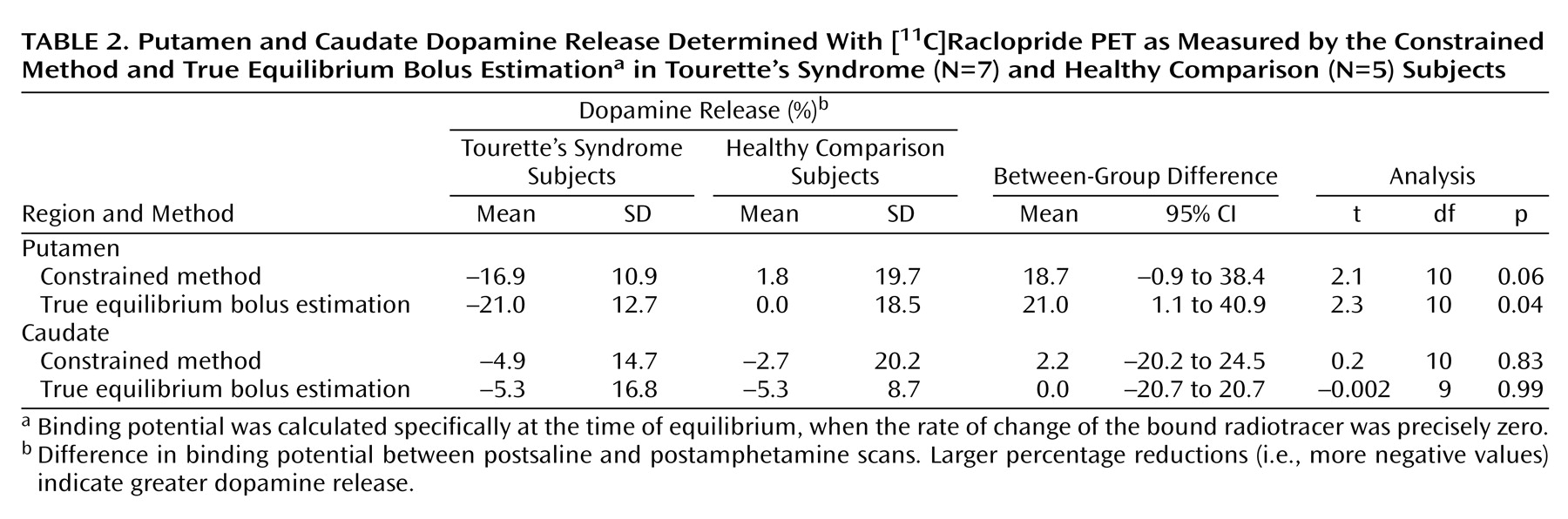
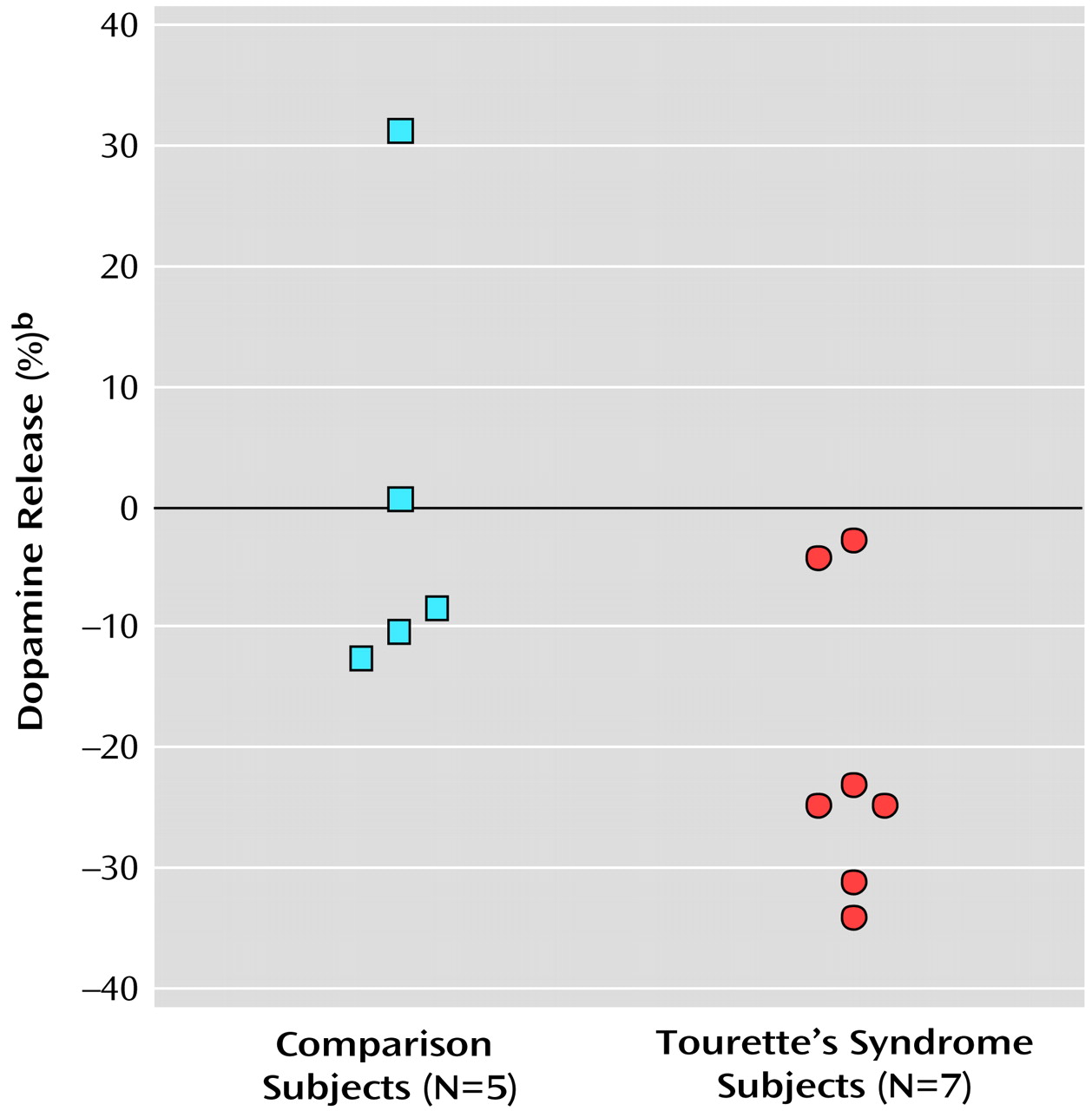
The present study suggests that after a stimulant challenge, there is a greater release of dopamine in the putamen of adults with Tourette’s syndrome relative to that seen in comparison subjects. Although the total number of subjects in our study is limited, the comparison group had dopamine release measures in the putamen consistent with prior reports that used an amphetamine [
123I]-IBZM SPECT paradigm
(24). In the putamen of Tourette’s syndrome subjects, values of intrasynaptic dopamine as measured by true equilibrium bolus estimation increased by 21% after amphetamine challenge, which was significantly larger than that seen in the comparison group. The difference in relative dopamine release in the putamen between Tourette’s syndrome and comparison subjects approached significance when the less sensitive constrained method of analysis was used. Although the change in dopamine release appears unrelated to age, tic severity, and OCD, whether it is associated with other symptoms, such as ADHD, remains unclear. Dopamine transporter density, measured by SPECT, has been shown to be greater in adults with ADHD than in healthy comparison subjects
(39). Although global tic severity ranking did not correlate with the degree of dopamine release, potential explanations include the small number of study subjects and characteristics relating to tics, i.e., their natural variability, possible improvement over time, and exacerbation by external factors such as stress, anxiety, fatigue, or the presence of infection
(40–
42).
Intrasynaptic release of dopamine in the caudate of Tourette’s syndrome subjects was not significantly different from that of the comparison subjects. It is possible that differences in the caudate were partially obscured by methodological issues—a greater effect of partial volume due to a smaller caudate area and the finite resolution of the PET scanner. It is also possible that, consistent with volumetric MRI studies that have shown changes in symmetry or size of the putamen but little alteration of the caudate
(43,
44), the putamen is the primary site of pathology in Tourette’s syndrome.
Although similar differences in putamen dopamine release were recorded when either true equilibrium bolus estimation or the constrained method of measurement was used, differences as measured by true equilibrium bolus estimation had greater significance. This is consistent with the belief that true equilibrium bolus estimation is a more optimal representation of binding potential and, in turn, the measurement of intrasynaptic dopamine release. In this relatively new method, binding potential and B
max, when used with a low specific activity injection, is calculated specifically at the time of equilibrium when the rate of change of the bound radiotracer is precisely zero following a bolus injection
(13,
33). Of the various methods for obtaining binding potential after a bolus injection, true equilibrium bolus estimation is considered the most appropriate.
Explanations for the higher intrasynaptic dopamine levels in the putamen of Tourette’s syndrome subjects following amphetamine stimulation are speculative. Among several possibilities are an increase in release of dopamine from the presynaptic terminal secondary to a localized defect in the release mechanism; a lack of presynaptic inhibition; an increase in firing of presynaptic neurons; or a functional defect in dopamine reuptake from the synaptic cleft. The modulation of dopamine release involves a complex interaction of reciprocal autoreceptor and heteroreceptor control
(45,
46). For example, the release of dopamine is inhibited by activation of α
2 adrenergic receptors
(47,
48) and facilitated by serotonin-1A receptors
(45). Furthermore, although levels of dopamine transporter binding may be affected in only some patients with Tourette’s syndrome
(15), a functional abnormality of dopamine reuptake from the synaptic cleft could elevate dopamine levels in synaptic vesicles, and, in turn, increase dopamine release.
An alternative unifying hypothesis could lie in the tonic-phasic model of dopamine release
(49). Based on electrophysiological and neurochemical data, two types of dopamine kinetics exist in the extracellular space. Tonic dopamine, which exists extracellularly in low concentration, determines the long-term or homeostatic mechanism. This “basal” level of dopamine is defined primarily as an extrasynaptic measure and calculated by use of microdialysis and electrophysiologic measurements. Dopamine autoreceptors (D
2 and D
3 subtypes) are proposed as regulators of tonic dopamine control
(46). Phasic dopamine is the spike-dependent dopamine released primarily into the synapse. It can escape the synaptic cleft with sufficient stimulation or when an uptake blocker is given in high concentrations. Intrasynaptic dopamine release induced by the use of stimulant challenges, such as with amphetamine, has been proposed as a surrogate measurement for phasic dopamine. The tonic-phasic model has been used to explain an increase in dopamine release observed in patients with schizophrenia
(24,
25,
49,
50).
Clinical and imaging studies, including our dopamine release data, are consistent with the possibility that the underlying pathobiology in Tourette’s syndrome is an abnormal regulation of the phasic dopamine response resulting in a hyperresponsive spike-dependent dopaminergic system. Two basic mechanisms are proposed for alteration of the phasic dopamine. The first is a lower sensitivity of phasic dopamine to tonic stimulation. This is unlikely, however, in view of the apparent marked responsiveness to a systemic dopamine agonist, as shown by the increase in dopamine release following our amphetamine challenge. A second possibility is a decrease in tonic dopamine levels. We favor this latter concept on the basis of lower levels of CSF HVA
(6,
8,
9) and evidence for elevated D
2 receptors in some subgroups of Tourette’s syndrome patients
(13). If this is indeed the case, how does the decrease in tonic dopamine occur? Diminished tonic dopamine levels could be secondary to a decrease in phasic overflow from the synaptic cleft to the extracellular space, but this is not likely because it conflicts with the observation of an increase in phasic dopamine release. A decrease in tonic dopamine could be secondary to a diminished cortical afferent input, i.e., tonic dopamine release is regulated by cortical glutamatergic afferents
(49). To our knowledge, however, postmortem and neuroimaging studies have not identified reduced cortical gray matter volume or abnormal efferent output. Anderson et al.
(51) evaluated postmortem 13 brain regions from four Tourette’s syndrome patients and reported lower glutamate levels in the globus pallidus and substantia nigra pars reticulata but not in the putamen. Last, a decrease in tonic dopamine levels could be secondary to an increase in activity of the dopamine transporter, since the reuptake transporter determines the concentration of extrasynaptic dopamine. Hence, we propose that the essential underlying mechanism in Tourette’s syndrome could be an overactive dopamine transporter system. This situation would create reduced levels of extracellular dopamine, higher concentrations of dopamine in the axon terminal, an increase in stimulus-dependent dopamine release, autoreceptor supersensitivity at the presynaptic site, and an increase in sensitivity to low-dose neuroleptics. Several clinical findings in Tourette’s syndrome patients support the overactive dopamine transporter hypothesis. For example, the exacerbation of tics by stimulant medications
(52,
53) could be secondary to greater dopamine release from the axon terminal. Environmental stimuli, such as stress, anxiety, and medications, well known to exacerbate tics, have been shown to increase phasic bursts of dopamine. Last, tic suppression with very low doses of neuroleptics
(54) may occur because there is less tonic dopamine available for the neuroleptic to block.
Future PET studies involving larger numbers of Tourette’s syndrome subjects and using techniques in which measurements of D2 receptors, dopamine transporter, and dopamine release are available for each subject should provide further clarification of the dopaminergic system in adults with Tourette’s syndrome. Confirmation of a release abnormality in larger numbers of patients with Tourette’s syndrome could, in turn, lead to the development of new tic-suppressing pharmacotherapies.