As we celebrate the 50th anniversary of Nobelists Watson, Crick, and Wilkin’s discovery (with Franklin) of the structure of DNA—and its offspring, the complete sequencing of the human genome—it is salutary to contemplate the relative youthfulness of the field of human genetics. The term “genetics” was provided by William Bateson in 1902 (the Wright brothers’ first flight was in 1903). In 1909, the clarifying distinction we now take for granted between the concept of “genotype” and the concept of “phenotype” was provided by the Danish botanist Wilhelm Johanssen. He also introduced the word “gene.” His research on self-fertilized lines of beans revealed that quantitative variability in the phenotype confounded thinking about separable contributions of heredity and environment. He found that the phenotype is often an imperfect indicator of the genotype, that the same genotype may give rise to a wide range of phenotypes, and that the same phenotype may have arisen from different genotypes
. Specific evidence for multifactorial (genetic and nongenetic) contributions to a continuous phenotype was provided about the same time by H. Nilsson-Ehle on the basis of observations of seed colors in crosses of oats and wheat. However, the term “polygene” was not available until K. Mather coined it in 1941. Exact citations for these historical references, often in German, are provided in the classic text by A.H. Sturtevant
(1).
Genotypes, which can be measured with techniques of molecular biology such as polymerase chain reaction (PCR) and DNA sequencing, are often useful as probabilistic prognosticators of disease. In contrast, a phenotype represents observable characteristics of an organism, which are the joint product of both genotypic
and environmental influences. In diseases with classic or Mendelian genetics as their distal causes, genotypes are usually indicative of phenotypes. However, this degree of genetic certainty does not exist for diseases with complex genetics
(2–
4). Genetic probabilism aptly describes the process by which a particular genotype gives rise to phenotype
(5,
6). Epigenetic factors may also be of critical importance for modifying the development of phenotypes
(7), and such modifications may be influenced by genotype or environment or be entirely stochastic in origin
(8). Thus, models of complex genetic disorders predict a ballet choreographed interactively over time among genotype, environment, and epigenetic factors, which gives rise to a particular phenotype
(9–
12).
Despite the successful characterization of the nucleotide base-pair order that represents the human genome
(13,
14), and although a legion of genetic linkage and association studies have been done, psychiatry has had little success in definitively identifying “culprit” genes or gene regions in the development of diseases categorized by using the field’s diagnostic classification schemas
(15–
18). The reason there is so much difficulty is undoubtedly—in part—that psychiatry’s classification systems describe heterogeneous disorders
(19–
22). In addition to the inherent complexity of psychiatric diseases, which have multifactorial and polygenic origins, the brain is the most complex of all organs. In organs such as the liver, all cells are nearly identical in their phenotypes and very similar in their transcriptomes (mRNA transcripts) and proteomes. In addition to the homogeneity in the structure of such cells, their interactions are mostly homogeneous. However, individual cells of the brain are quite different from each other in their transcriptomes, proteomes, and morphological phenotypes and also in the thousands of connections and interactions with other neurons and glia that are critically important to optimal functioning. Different cellular experiences are transduced to differences on the biochemical and epigenetic levels so that cellular memories regulated by protein modification, morphometric changes, and epigenetic influences make the brain unique among organs. Furthermore, the brain is subject to complex interactions not just among genes, proteins, cells, and circuits of cells but also between individuals and their changing experiences
(23). Therefore, the phenotypic output from the brain, i.e., behavior, is not simply a sum of all its parts. It stands to reason that more optimally reduced measures of neuropsychiatric functioning should be more useful than behavioral “macros” in studies pursuing the biological and genetic components of psychiatric disorders.
The Endophenotype Concept in Psychiatry
The theory that genes and environment combine to confer susceptibility to the development of diseases surfaced in the early half of the last century, but the use of such a framework for exploring the etiology of schizophrenia and other psychiatric disorders is more recent. Douglas Falconer’s 1965 multifactorial threshold model for diabetes and other common, non-Mendelizing diseases was adapted to a polygenic model of schizophrenia in 1967
(24). About this time, it became clear that the classification of psychiatric diseases on the basis of overt phenotypes (syndromic behaviors) might not be optimal for genetic dissection of these diseases, which have complex genetic underpinnings. In their writings summarizing genetic theories in schizophrenia 30 years ago, Gottesman and Shields
(25,
26) described “endophenotypes” as internal phenotypes discoverable by a “biochemical test or microscopic examination.” The term was adapted from a 1966 paper by John and Lewis
(27), who had used it to explain concepts in evolution and insect biology. They wrote that the geographical distribution of grasshoppers was a function of some feature not apparent in their “exophenotypes”; this feature was “the endophenotype, not the obvious and external but the microscopic and internal.”
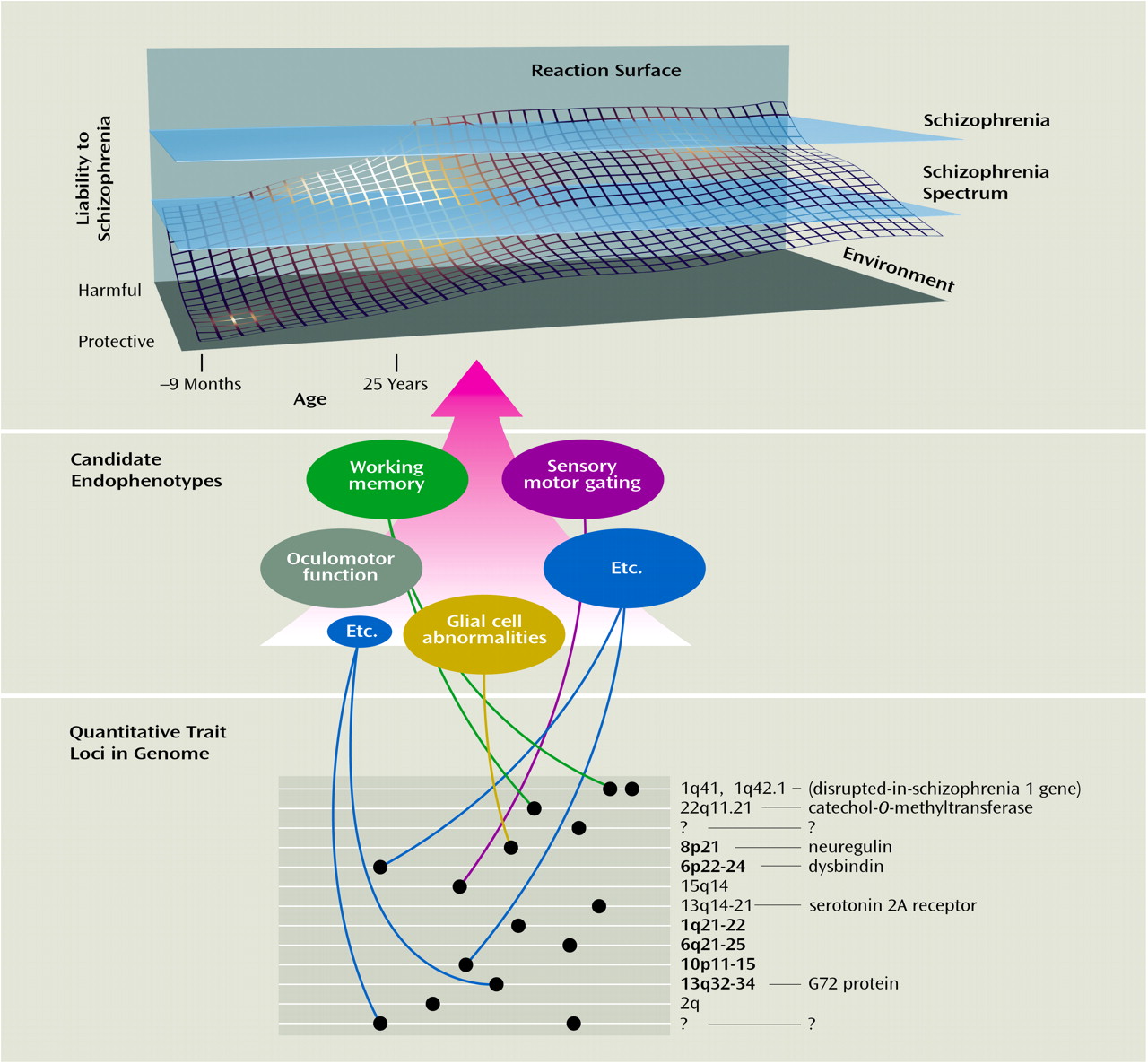
That felicitous term seemed to suit the needs of psychiatric genetics, and the concept of endophenotype was adapted for filling the gap between available descriptors and between the gene and the elusive disease processes. The identification of endophenotypes, which do not depend on what was obvious to the unaided eye, could help to resolve questions about etiological models. The rationale for the use of endophenotypes in exploring disease processes is illustrated in
Figure 1. This rationale held that if the phenotypes associated with a disorder are very specialized and represent relatively straightforward and
putatively more elementary phenomena (as opposed to behavioral macros), the number of genes required to produce variations in these traits may be fewer than those involved in producing a psychiatric diagnostic entity. Endophenotypes provided a means for identifying the “downstream” traits or facets of clinical phenotypes, as well as the “upstream” consequences of genes and, in principle, could assist in the identification of aberrant genes in the hypothesized polygenic systems conferring vulnerabilities to disorders. That is, the intervening variables or hypothetical constructs that were championed as useful for theorizing about behaviors
(35)—and that could mark the path between the genotype and the behavior of interest (
Figure 2)—might Mendelize in a predicted manner.
Despite the inherent advantages of the concept of endophenotype, the term and its promise lay dormant for a number of years. However, now that multiple genetic linkage and association studies using current classification systems and the development of practical animal models, have all fallen short of success, the term and its usefulness have reemerged. (A MEDLINE search for the years 2000 through 2002 found 62 entries for “endophenotype,” compared with 16 entries before 2000.) Endophenotypes are being seen as a viable and perhaps necessary mechanism for overcoming the barriers to progress
(28,
51–58). The methods available for endophenotype analysis have advanced considerably since 1972; our current armamentarium includes neurophysiological, biochemical, endocrinological, neuroanatomical, cognitive, and neuropsychological (including configured self-report data) measures
(29). Advanced tools of neuroimaging such as functional magnetic resonance imaging (fMRI), morphometric MRI, diffusion tensor imaging, single photon emission computed tomography (SPECT), and positron emission tomography (PET) promise to expand the possibilities even more
(30,
59–61). Other terms with patently synonymous meaning, such as “intermediate phenotype,” “biological marker,” “subclinical trait,” and “vulnerability marker,” have been used interchangeably. These terms may not necessarily reflect genetic underpinnings but may rather reflect associated findings (see the discussion in the next section). In this context, we use the term “biological marker” to signify differences that do not have genetic underpinnings and “endophenotype” when certain heritability indicators are fulfilled.
Endophenotypes in Genetic Analysis
An endophenotype-based approach has the potential to assist in the genetic dissection of psychiatric diseases. Endophenotypes would ideally have monogenic roots; however, it is likely that many would have polygenic bases themselves. Furthermore, the use of endophenotypes in genetic research must be tempered by the realization that without controls and limits, their usefulness may be obscured. For example, putative endophenotypes do not necessarily reflect genetic effects. Indeed, these biological markers may be environmental, epigenetic, or multifactorial in origin. Criteria useful for the identification of markers in psychiatric genetics have been suggested
(62) and have been adapted here to apply to endophenotypes:
1. The endophenotype is associated with illness in the population.
2. The endophenotype is heritable.
3. The endophenotype is primarily state-independent (manifests in an individual whether or not illness is active).
4. Within families, endophenotype and illness co-segregate.
Subsequently, an additional criterion that may be useful for identifying endophenotypes of diseases that display complex inheritance patterns was suggested
(29):
5. The endophenotype found in affected family members is found in nonaffected family members at a higher rate than in the general population.
Other fields of medicine have had some success in using endophenotypes to assist with genetic linkage studies. For instance, the multiple genes that cause long QT syndrome were identified by using an endophenotype-based method
(63,
64). Manifestations of long QT syndrome include syncope, ventricle arrhythmias, and sudden death
(63). Although not all family members who carry the disease genes show these symptoms, a much greater percentage have QT elongation as measured by ECG. By using QT elongation as a phenotype—and excluding or including pedigree members with this finding—linkage studies were successful in identifying the genes that cause the QT elongation endophenotype and thus the syndrome phenotypes of syncope, ventricle arrhythmias, and sudden death
(64,
65). The identification of these genes has allowed for genetic manipulations in mice to study disease pathology and to further the development of novel medications
(66). Other examples in the literature of endophenotype-based strategies for identifying genetic linkage include studies of idiopathic hemochromatosis (excessive serum iron)
(67), juvenile myoclonic epilepsy (an EEG abnormality)
(68), and familial adenomatous polyposis coli (intestinal polyps)
(69). In other disorders with complex genetics such as diabetes, hypercholesterolemia, or hypertension, researchers use physiological challenges, biochemical assays, and physiological measures to obtain a primary index of disease pathology. Indeed, these syndromes may all present to the physician as fatigue, but the pathophysiological underpinnings are substantially different. The glucose tolerance test, measurements of serum cholesterol levels, and sphygmomanometer measurements all represent objective, quantifiable methods for making disease diagnosis and classification. In addition to being crucial in diagnosis and classification of these diseases, the phenomena measured by these methods constitute endophenotypes that represent the
primary inclusion/exclusion feature by which “hits” for genetic linkage and association studies are defined.
In psychiatry, a number of attempts have been made to develop and determine the feasibility of candidate endophenotypes. However, few have met all the criteria listed earlier. Nonetheless, some linkage and association studies—using endophenotypes—have had moderate success. Candidate endophenotypes have also been used in the development of animal models and to subtype patients for classification and diagnostic reasons (see the discussion in later sections). The hunt for candidate endophenotypes has been described in the literature on several psychiatric disorders, including schizophrenia
(30,
31–
33,
39,
70–73), mood disorders
(28,
55,
74,
75), Alzheimer’s disease
(76,
77), attention deficit hyperactivity disorder
(54,
78,
79), and even personality disorders
(80). We give a brief description of some possibilities in schizophrenia research as salient examples. The interested reader is referred to the references just cited for more in-depth discussions.
Sensory Motor Gating and Eye-Tracking Dysfunction in Schizophrenia
Deficits in sensory motor gating are consistent neuropsychological findings in schizophrenia
(33,
39). The hypothesized association between these deficits and schizophrenia has face validity primarily on the basis of patients’ reports that they have difficulty filtering information from multiple sources
(33,
81–83). On the level of neurobiology, the inhibitory mechanisms of patients with schizophrenia may not be capable of adequately adjusting to the multiple distinct or repetitive inputs that occur in everyday life. Neuropsychological tests, including assessments of P50 suppression and prepulse inhibition of the startle response, have been developed to discern efficiencies in these capabilities. Both tasks have been studied in schizophrenic patients, and abnormalities consistent with defects in inhibitory neuronal circuits have been found.
In tests of prepulse inhibition, startling sensory stimuli (loud noise, bright light) are used to elicit an unconditional reflexive startle response in individuals. If a weaker prestimulus is provided before the startling stimulus, the subsequent startle response is generally diminished. A relatively reproducible finding is that this dimunition of the second response is attenuated in patients with schizophrenia, compared to healthy subjects
(39,
84,
85). Prepulse inhibition is a generally conserved finding among vertebrates, and as such it has been the target of several rodent studies (reviewed in reference
86), both to model a facet of schizophrenia and to investigate the biology of a prepulse inhibition response. The presence of this candidate endophenotype has been documented in relatives of patients with schizophrenia
(87), but more extensive testing is required. Genetic studies in inbred animals have suggested at least a partial genetic diathesis
(86); however, environmental influences may also be active
(88,
89). Abnormal prepulse inhibition is not specific to schizophrenia; studies have identified this abnormality in obsessive-compulsive disorder
(90) and Huntington’s disease
(91), among others. However, the reproducibility of the finding in schizophrenia, the fact that abnormal prepulse inhibition parallels a putative central abnormality in the disease, and the fact that prepulse inhibition is a conserved phenomenon among vertebrates make abnormal prepulse inhibition a promising candidate endophenotype to pursue.
The P50 suppression test uses two auditory stimuli presented at 500-msec intervals. A positive event-related response for both stimuli is measured by EEG. In normal individuals, the neuronal response to the second stimulus is of lower amplitude than the first. However, patients with schizophrenia do not show the same degree of suppression of P50 amplitude
(33,
92–95). In addition to this finding in probands, abnormal P50 suppression is found in unaffected first-degree relatives of patients with schizophrenia
(95–
99). The heritability of this measure has been assessed in twins, and the results have suggested that genetics plays a role in the development of variation in this candidate endophenotype
(100,
101). Freedman and colleagues
(102) also used P50 suppression to identify a potential susceptibility locus for schizophrenia on chromosome 15, a chromosomal region where the gene for the α7 nicotinic acetylcholine receptor resides. Furthermore, this group of researchers has shown linkage disequilibrium in this region
(103) and has shown that promoter variants of the α7 receptor are associated with schizophrenia and/or P50 suppression abnormalities
(104).
Eye-tracking dysfunction has long been associated with schizophrenia. This dysfunction was first described in 1908 by Diefendorf and Dodge
(105), whose work was rediscovered in the 1970s, initially by Holzman and colleagues
(106,
107).
Eye movements are generally of two forms, either saccadic (brief and extremely rapid movements) or smooth and controlled. The latter “smooth pursuit” eye movements occur only when the subject is following an object moving at a constant velocity, most commonly a pendulum (in early studies) or bright dot on a computer monitor. Initiation and maintenance of smooth pursuit eye movements involve integration of functions of the prefrontal cortex frontal eye fields, visual and vestibular circuitry, thalamus, and cerebellum, as well as the muscles and neural circuitry directly responsible for eye movement
(108).
A number of studies have found that patients with schizophrenia have deficiencies in smooth pursuit eye movements, compared to healthy subjects (see references
41–
43 for review). In general, these deficiencies are manifested as corrective saccades, which follow smooth pursuit eye movements that are slightly slower than the target (reviewed in reference
42, where more detailed descriptions of specific abnormalities are available). Furthermore, the heritability of these deficiencies has been extensively addressed; studies have suggested that biological relatives of schizophrenic subjects have an increased rate of smooth pursuit eye movement dysfunction. Thus, 40%–80% of schizophrenic subjects, 25%–45% of their first-degree relatives, and less than 10% of healthy comparison subjects generally show this trait
(41–
43). A study requiring replication has suggested linkage to a region of chromosome 6
(109). Correlating smooth pursuit function with neuroimaging measures
(110) or performance on working memory tasks
(111,
112) may be a useful research strategy. Smooth pursuit eye movements are maintained in primates but not in most other mammals used in preclinical research
(108).
Working Memory in Schizophrenia
Working memory and executive cognition are compromised in patients with schizophrenia
(44). A primary brain region involved in working memory is the dorsolateral prefrontal cortex
(31,
45,
113), a region in which abnormalities have been found in postmortem studies of schizophrenic patients
(114). Family
(115,
116), and twin studies
(117,
118) have suggested heritability of working memory deficits in schizophrenia.
Recent studies have identified gene and chromosomal regions possibly involved in working memory. A study of Finnish twins by Gasperoni and colleagues
(53), which used an endophenotype-based strategy, suggested linkage and association to a region of chromosome 1. In their study, dizygotic twins discordant for schizophrenia underwent four neuropsychological tests. Using the sum of performance scores on these tests, Gasperoni and colleagues identified significant linkage to 1q41, a region previously suggested in traditional linkage studies of schizophrenia
(119–
122). By stratifying their data according to performance on each neuropsychological test, they found that visual working memory performance was highly significantly linked with this region (p=0.007), while performance on none of the other three neuropsychological tests was significantly associated with any 1q markers. In the second part of their study, Gasperoni and colleagues
(53) completed an association analysis involving monozygotic discordant twins, unaffected dizygotic and monozygotic twins, and the dizygotic twin group from the linkage study. In this analysis, an association of the 1q41 region and performance on the visual working memory task was again identified. The facts that previous linkage studies have identified this region
and that performance on working memory tasks is a reproducible endophenotype for schizophrenia strengthen the claim that this endophenotype—and the putative gene(s) at 1q41 linked to it—may be relevant to the pathophysiology of schizophrenia. The study requires replication in a larger group of subjects representing a nonisolate population.
Association and physiological evidence have also linked a specific enzyme with a small increased risk for developing schizophrenia and with poorer performance on a working memory task. The enzyme catechol
O-methyltransferase (COMT), the gene for which is found at 22q11.2, assists in the catabolism of dopamine. This chromosomal region has been linked to both schizophrenia and bipolar disorder and overlaps with a deletion that has been associated with velocardiofacial syndrome (DiGeorge syndrome) and schizophrenia (see reference
16 for review). A functional polymorphism (
val108/158met) for COMT results in a fourfold increase in the activity of this enzyme. The considerable body of evidence implicating dopaminergic neurotransmission, the presence of a common functional polymorphism, and the data suggesting the involvement of the dorsolateral prefrontal cortex in schizophrenia and working memory led to association studies of COMT
(31).
While their effect sizes are small, a number of family studies have found that the valine allele is transmitted at a higher rate than the methionine allele to patients with schizophrenia than to their nonaffected siblings (reviewed in reference
31). This polymorphism has also been linked to performance on a working memory task. Specifically, Egan et al.
(123) associated poorer performance on a working memory task in patients, their siblings, and comparison subjects with the same valine allele variation of COMT found to be transmitted at a higher rate in schizophrenia. They used fMRI to measure dorsolateral prefrontal cortex activation in a subset of these individuals; the fMRI fingerprint from individuals with the valine allele suggested that activation of the dorsolateral prefrontal cortex is less efficient in those subjects
(123). Additional studies from two independent laboratories have also suggested that patients with schizophrenia show this inefficiency
(124–
126). Callicott and colleagues
(127) have recently shown that the fMRI response in the dorsolateral prefrontal cortex observed in schizophrenic subjects is also found in unaffected siblings of patients with schizophrenia. Although they found no group differences between the siblings of schizophrenic patients and the comparison group in overall working memory performance, fMRI measurement showed that the sibling group had less efficient dorsolateral prefrontal cortex functioning than the comparison group. Taken together, these results suggest that fMRI analysis of subjects undergoing working memory tasks may be a more sensitive endophenotype than working memory performance alone as measured by neuropsychological testing. Additional studies using PET have suggested dysfunction of the cortical-thalamic-cerebellar-cortical circuit during working memory tasks
(72,
73). The “cognitive dysmetria” resulting from this disruption may provide another candidate endophenotype.
Conclusions: Broader Uses for Endophenotypes
Endophenotypes may have additional uses in psychiatry, including uses in diagnosis, classification, and the development of animal models. The current classification schema in psychiatry were derived from observable clinical grounds to address the need for clinical description and communication
(22). However, they are not based on measures of the underlying genetic or biological pathophysiology of the disorders. The most widely used systems currently in place must serve the needs of clinicians, psychiatric statisticians, administrators, and insurance companies, among other groups and agencies
(128). As this system is designed for a wide range of users and because it pays little attention to the biological contributors to the disorders, it is not optimized for the design, implementation, and success of research studies
(128). The lack of a biological basis for the classification of psychiatric disorders has led, in part, to a lack of success in studies of the neurobiology and genetics of psychiatric disorders. Endophenotype-based analysis would be useful for establishing a biological underpinning for diagnosis and classification; a net outcome would be improved understanding of the neurobiology and genetics of psychopathology.
Animal models are an active area of research in psychiatry. However, despite some progress
(129,
130), there remains a great need for further development
(130–
132). Improved animal models will help in understanding the neurobiology of psychiatric disorders and will further the development of truly novel medications
(133). Development of animal partial-models in psychiatry relies on identifying critical components of behavior (or other neurobiological traits) that are representative of more complex phenomena
(134). Animals will never have guilty ruminations, suicidal thoughts, or rapid speech. Thus, animal models based on endophenotypes that represent evolutionarily selected and quantifiable traits may better lend themselves to investigation of psychiatric phenomena than models based on face-valid diagnostic phenotypes
(28).
Given the hopefully successful consequences of studies adopting an endophenotype strategy, psychiatric diagnosis will continue to be important in research and clinical practice. Indeed, similar to the principle we describe here, optimally reduced or partitioned phenotypes may be useful in refining the diagnostic system. Measures that have already been used to deconstruct illnesses for genetic analysis include severity and course of illness
(135), age at onset of illness
(136,
137), amount of substance use in drug and alcohol disorders
(138,
139), and response to specific treatments such as lithium
(140,
141).
Gottesman and Shields
(25) concluded their 1972 book on schizophrenia and genetics with the following remarks:
We are optimistically hopeful that the current mass of research on families of schizophrenics will discover an endophenotype, either biological or behavioral (psychometric pattern),which will not only discriminate schizophrenics from other psychotics, but will also be found in all the identical co-twins of schizophrenics whether concordant or discordant. All genetic theorizing will benefit from the development of such an indicator (p. 336).
Although these words are still pertinent after 30 years, there is ample reason to be optimistic about anticipated discoveries and refinements in the quest for endophenotypes.