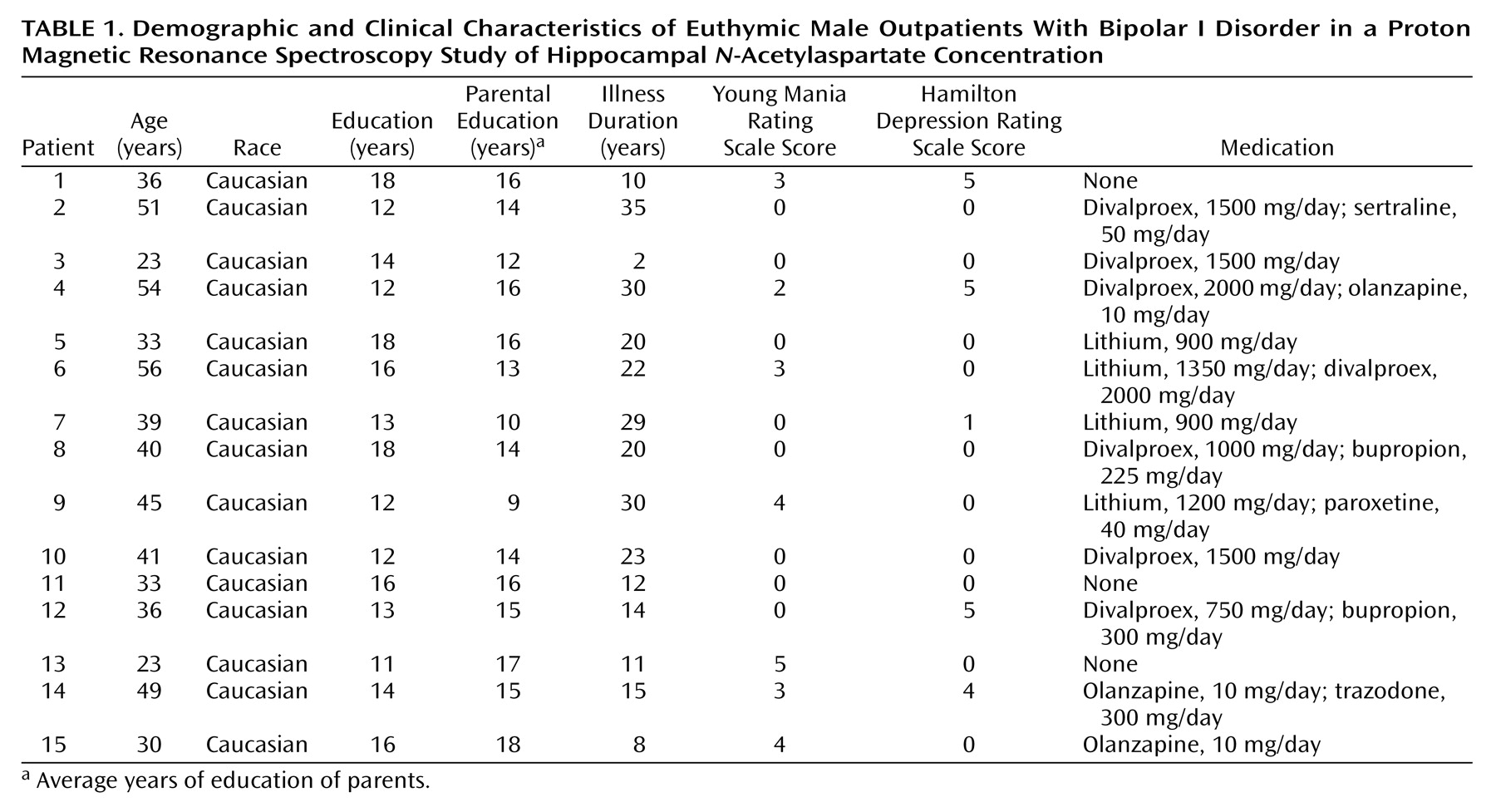
Evidence has been far from conclusive as to whether there are abnormalities of hippocampal structure and function in bipolar disorder. Whereas a smaller total number and smaller density and size of nonpyramidal hippocampal neurons have been reported by Benes and colleagues
(1), six of the seven published volumetric magnetic resonance imaging (MRI) studies of bipolar disorder that we are aware of reported no differences in hippocampal volumes of bipolar disorder patients relative to comparison subjects
(2–
7). Only one study noted a smaller right hippocampus in the bipolar disorder group
(8). However, three high-resolution MRI studies of recurrent major depression demonstrated significantly smaller hippocampal volumes
(9–
11). Furthermore, in one study, smaller hippocampal volumes were correlated with the longer duration of major depression, suggesting that recurrent episodes of depression may result in hippocampal neuronal loss
(9). One question that arises from these studies of mood disorders is whether hippocampal pathology in bipolar disorder is more subtle than that observed in major depression, perhaps too subtle to be reliably detected by MRI volumetric techniques. Furthermore, MRI volumetric techniques are currently not able to differentiate between smaller gray matter tissue volumes resulting from neuronal loss or atrophy and smaller volumes resulting from reductions in glial cell number or density.
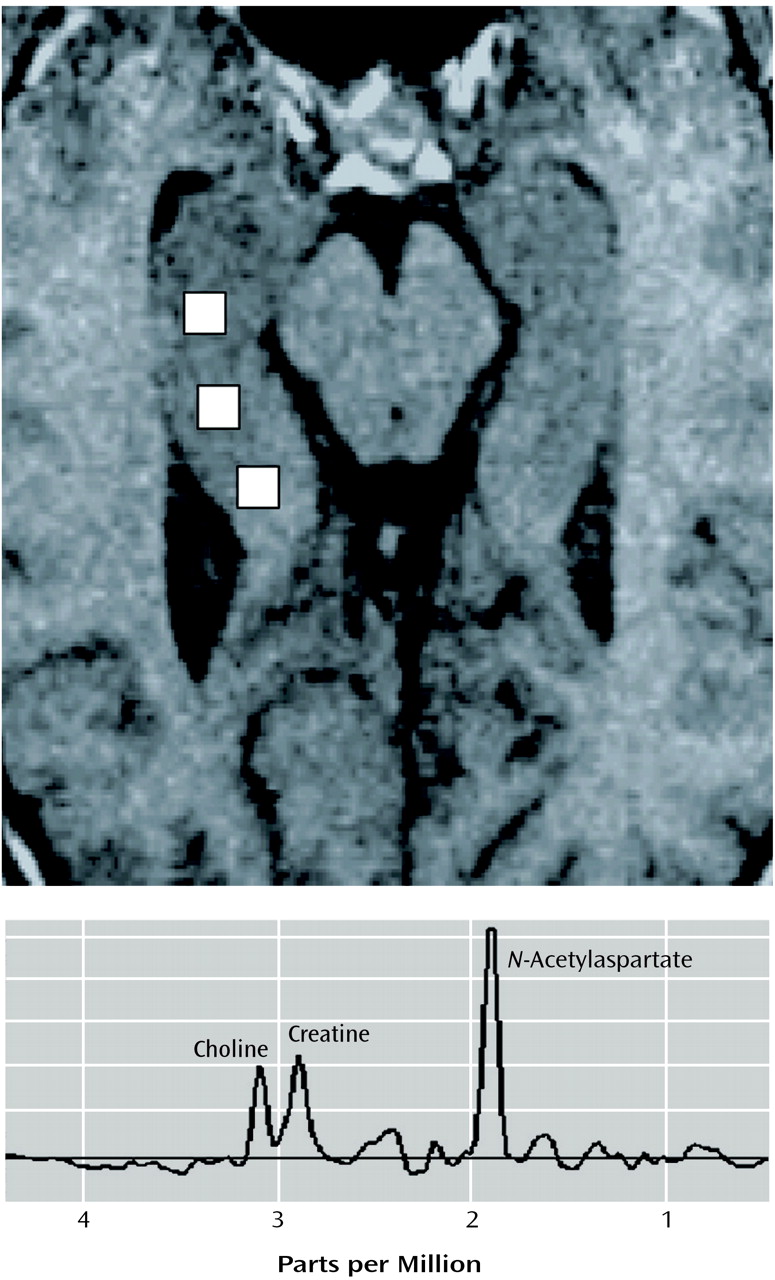
N-Acetylaspartate is the second most abundant amino acid in the central nervous system and is reliably measured by proton magnetic resonance spectroscopy (
1H-MRS).
N-Acetylaspartate is formed in mitochondria from acetyl coenzyme A and aspartate by the membrane-bound enzyme
l-aspartate
N-acetyltransferase, an enzyme selectively found in brain
(12). In studies using immunohistochemical techniques,
N-acetylaspartate has been shown to be predominantly localized to neurons, axons, and dendrites within the CNS
(13). Animal models of neuronal injury have been shown to give good correlations between
N-acetylaspartate levels as measured by
1H-MRS and neuronal counts as measured by histological techniques
(14–
16). Low
N-acetylaspartate is thought to represent loss of neurons and/or axons, reduction of interneuronal neuropil, and neuronal or axonal metabolic dysfunction or damage
(17–
20). Our previous study of patients with schizophrenia
(21) found low
N-acetylaspartate bilaterally in the absence of smaller hippocampal volume as measured by MRI, supporting the idea that
N-acetylaspartate may be a more sensitive marker of neuronal damage or loss than quantitative MRI measurements of tissue loss.
The choline signal measured by
1H-MRS is derived predominantly from constituents of membrane phospholipid metabolism
(22,
23), such as glycerophosphocholine and phosphocholine, which account for more than 50% of the resonance, and glycerophosphoethanolamine and phosphoethanolamine, which account for between 10% (in adults) and 25% (in the newborn) of the resonance. The choline signal is known to be significantly higher in conditions where there is ongoing myelin breakdown and/or glial cell proliferation, such as multiple sclerosis, adrenoleukodystrophy, brain tumors, and reactive gliosis
(24,
25). While two earlier studies
(26,
27) reported that changes in dietary choline could affect the choline signal, more recent studies have demonstrated that neither short-term nor long-term administration of oral choline appears to alter the
1H-MRS choline resonance or the proton-decoupled phosphorous MRS glycerophosphorylcholine and phosphorylcholine resonances in cortical gray matter, cortical white matter, basal ganglia, thalamus, and cerebellum
(28,
29).
To our knowledge, no published studies to date have used
1H-MRS to investigate hippocampal neuronal loss or dysfunction in bipolar disorder. Renshaw and colleagues
(30) reported lower
N-acetylaspartate measures bilaterally in the temporal lobe in 13 patients with first-episode psychosis, including six bipolar disorder patients; however, their findings were not specific to the hippocampus. Therefore, we used
1H-MRS to test the hypothesis that hippocampal
N-acetylaspartate was lower in patients with bipolar I disorder, suggesting neuronal loss and/or dysfunction. A second objective was to determine if the choline level in this region was higher, suggesting possible myelin breakdown or significant glial cell proliferation. A third objective was to determine if lower measures of
N-acetylaspartate in the hippocampus were associated with illness duration. To avoid the potential confounding effects of gender and clinical-state-dependent changes on
N-acetylaspartate measures, and because emerging evidence has suggested that abnormalities in brain structure and function may be more prominent in familial mood disorders
(31,
32), this study examined only euthymic male patients with familial bipolar I disorder.
Discussion
To our knowledge, this is the first published report of significantly lower
N-acetylaspartate in both the right and left hippocampus in euthymic male patients with familial bipolar I disorder, relative to healthy comparison subjects. Previous
1H-MRS studies of other brain regions in bipolar disorder have reported lower
N-acetylaspartate bilaterally in the dorsolateral prefrontal region
(46), as well as normal
N-acetylaspartate measures in the lenticular nuclei
(47) and frontal lobes
(48). Given that
N-acetylaspartate is found only in neurons and axons but not mature glial cells, lower hippocampal
N-acetylaspartate measures suggest loss of neurons and/or axons, reduction of interneuronal neuropil, neuronal or axonal metabolic dysfunction, or some combination of these processes. The finding of normal choline levels in the bipolar I disorder patients suggests that there is no biochemical evidence of myelin breakdown or glial cell proliferation in the hippocampus. A finding of lower hippocampal
N-acetylaspartate is consistent with the smaller total number, cell density, and cell size of nonpyramidal hippocampal neurons observed in the postmortem study by Benes and colleagues
(1).
Lower hippocampal
N-acetylaspartate may be the result of stress-induced cellular changes mediated by higher levels of glucocorticoids. The two major structural changes in the hippocampus that occur as a result of stress are atrophy of CA3 pyramidal neurons
(49) and reduced adult neurogenesis of granule cells in the dentate gyrus
(50,
51). A number of studies over the years have demonstrated how stress and adrenal glucocorticoids directly cause hippocampal neuronal atrophy as well as reduce cellular resilience, thereby making neurons more vulnerable to ischemia, hypoglycemia, and excitatory amino acid toxicity
(49,
52,
53). Since hypercortisolemia is a frequent finding during episodes of major depression, elevations in glucocorticoid levels may induce similar hippocampal neuronal alterations as a result of repeated episodes of depression; however, the evidence for this relationship at present remains indirect
(53). Therefore, it is reasonable to hypothesize that glucocorticoid-induced cell damage or loss, thought to be responsible for the smaller hippocampal volumes observed by means of MRI in major depression, may also be occurring in bipolar I disorder, resulting in compromised neuronal function or loss and lower hippocampal
N-acetylaspartate measures. However, it should be noted that none of the bipolar I disorder patients in this study were assessed for elevations in glucocorticoids.
Alterations in neurotrophic or neuroprotective factors may also underlie lower hippocampal
N-acetylaspartate measures. Stress appears to decrease expression of brain-derived neurotrophic factor (BDNF)
(54), which is critical for both the survival and function of neurons in the developing and adult brain
(55). Thus, stress-induced reductions in BDNF could lead to damage and loss of hippocampal neurons. Another major neuroprotective and neurotrophic factor, B-cell lymphoma/leukemia-2 gene proteins (Bcl-2), inhibits programmed cell death
(56,
57); yet, when overexpressed, Bcl-2 promotes regeneration of axons and regulates neurite sprouting
(58). It is interesting to note that lithium and valproate appear to robustly increase Bcl-2 levels in both in vitro and in vivo experimental models
(20). Both BDNF and Bcl-2 expression are regulated by the transcription factor cyclic adenosine monophosphate response element-binding protein (CREB), and recent studies have shown lower levels of CREB in the temporal cortex in depressed patients
(59) and up-regulated CREB and hippocampal expression of BDNF in response to chronic antidepressant treatment
(60–
62). Findings such as these support the idea that major mood disorders could result from a failure or compromise of mechanisms that regulate neuronal plasticity. It is therefore conceivable that reductions in BDNF and/or Bcl-2 are occurring in bipolar I disorder, adversely affecting hippocampal neuronal plasticity and survival and resulting in a lower hippocampal
N-acetylaspartate levels.
Alternatively, a lower hippocampal
N-acetylaspartate may be a consequence of alterations in reelin, a secretory glycoprotein responsible for normal lamination of the brain. In the adult mammalian brain, reelin is localized to layer I cortical Cajal-Retzius cells, cortical and hippocampal interneurons, and cerebellar granule cells
(63,
64). Reelin contributes to neuronal plasticity in the brains of adults by acting on integrin receptors expressed in spine postsynaptic densities. An important consequence of reelin signaling is the activation of the focal adhesion tyrosine kinase system, which is a component of a postsynaptic mechanism responsible for an increase in the number of synapses and alterations in postsynaptic structure in axons, dendrites, and the intermediate filament cytoskeleton of astrocytes
(63,
65). The heterozygous reeler mouse, which expresses only 50% of the reelin protein levels seen in wild-type mice, exhibits reduced cortical neuropil expression, neuronal packing density, and dendritic spine density
(66). Recent studies of patients with bipolar disorder have reported 1) lower blood levels of reelin and its isoforms
(63), 2) lower reelin mRNA in prefrontal cortex and cerebellum
(64,
67), and 3) lower reelin protein in the CA4 areas of the hippocampus
(68). Therefore, lower levels of hippocampal reelin in bipolar disorder could result in lower hippocampal
N-acetylaspartate, reflecting shrinkage of the interneuronal neuropil.
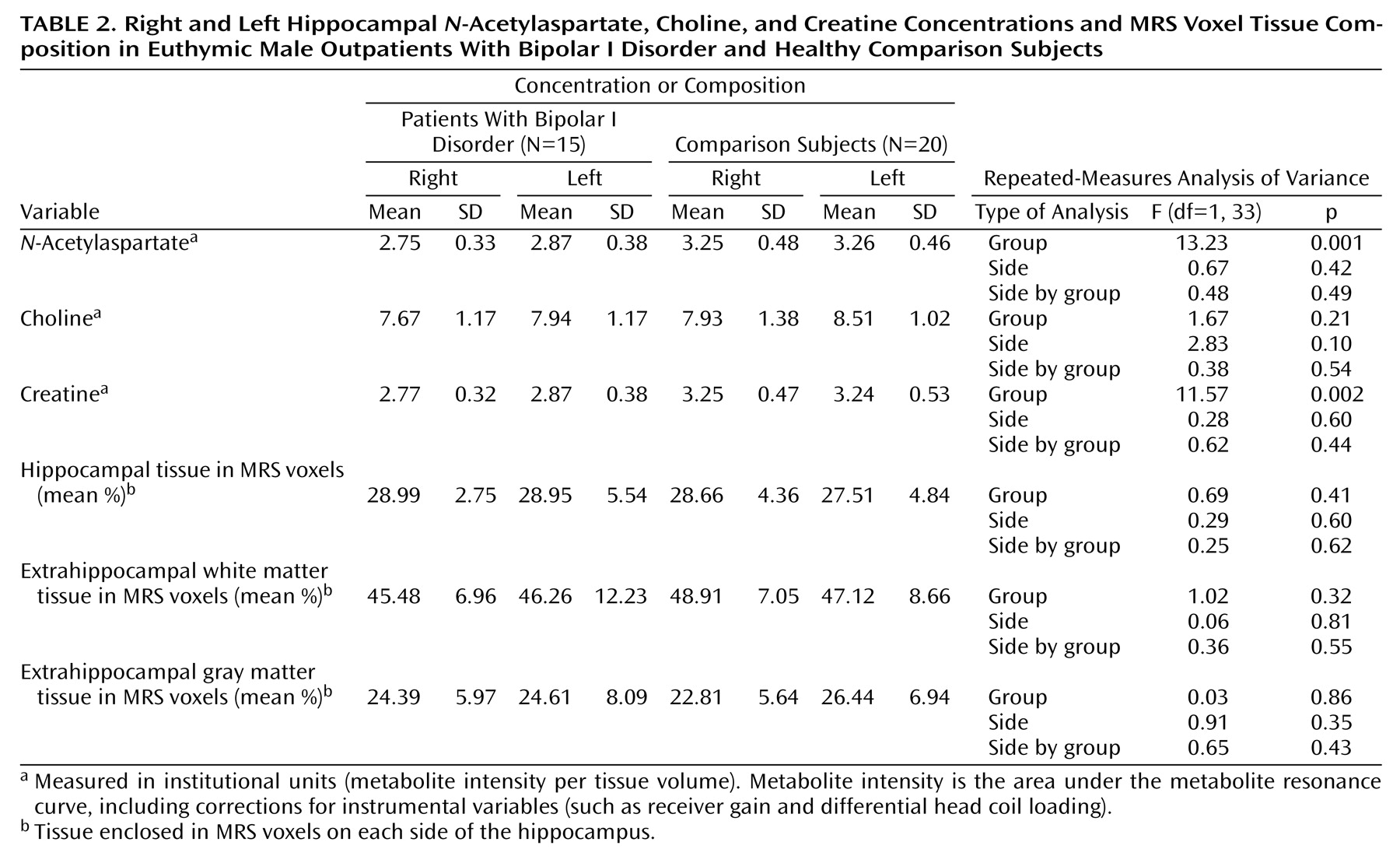
The second finding of this study was a lower concentration of hippocampal creatine bilaterally in the bipolar I disorder patients than in the comparison subjects. The creatine signal measures both creatine and phosphocreatine together, which are in rapid chemical enzymatic exchange in the human brain. In addition, the level of creatine in the brain can be significantly altered by osmotic forces as well as by extracerebral events due to the complex biosynthetic pathway through liver and kidney enzymes
(24). Because of this complexity, it is difficult to attribute differences in creatine simply to local derangements of cellular energy metabolism. Nevertheless, since phosphocreatine is linked to ATP through the creatine-kinase equilibrium, alterations in creatine could be related to changes in hippocampal metabolism.
The third finding of this study was that lower hippocampal N-acetylaspartate and creatine concentrations cannot be attributed to differences in voxel tissue heterogeneity. This analysis was made possible by using information from coregistered MRI tissue segmentation data. This result is important because it documents that the observed alterations in hippocampal N-acetylaspartate and creatine are not merely a reflection of differences in the tissue composition of the MRS voxels. Furthermore, it emphasizes the value of using MRI-derived tissue segmentation information to improve the analysis of spectral metabolite data from 1H-MRS investigations.
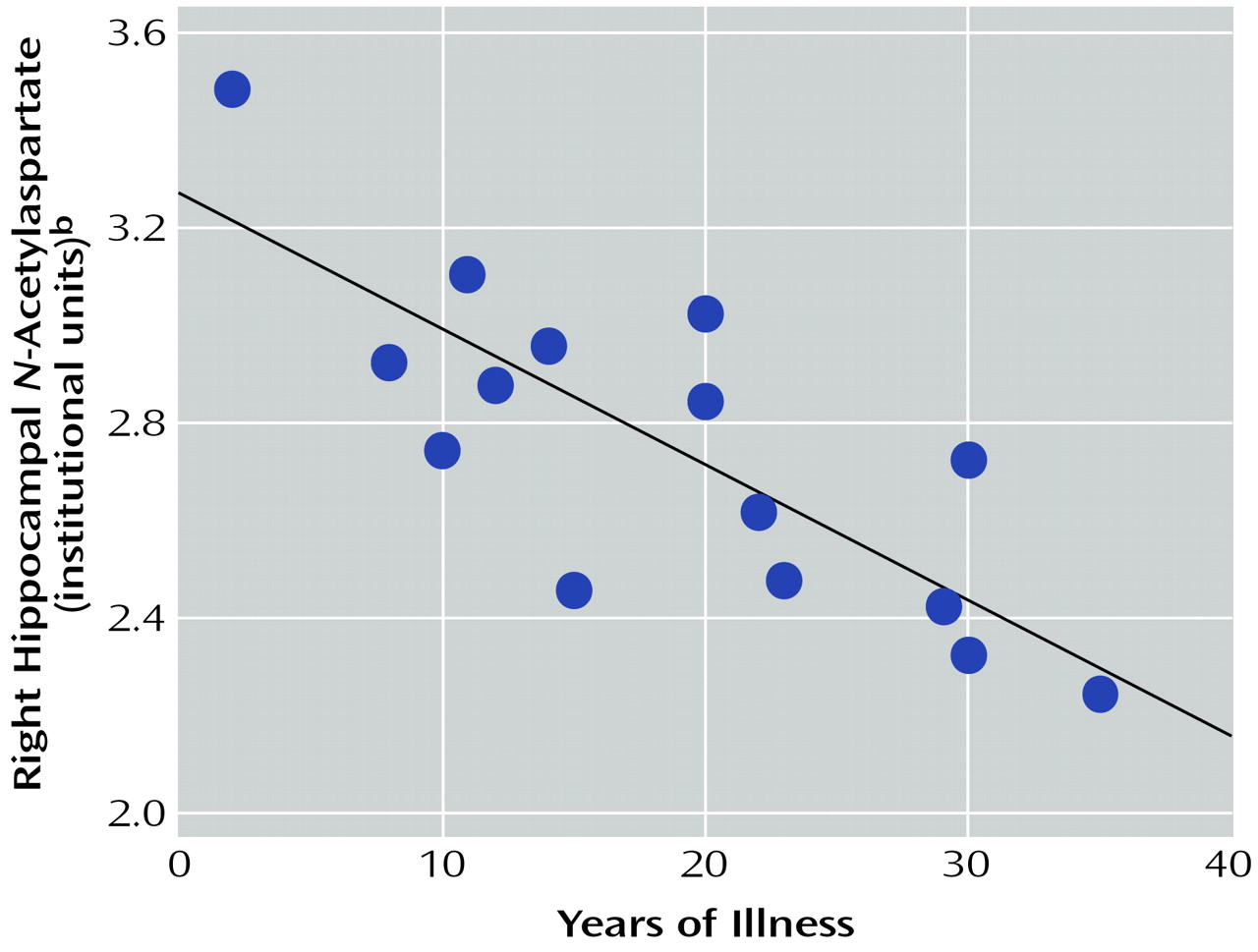
The fourth finding of this study was the presence of a significant negative correlation between right hippocampal
N-acetylaspartate concentration and illness duration. This finding supports the idea that neuronal atrophy, loss, or dysfunction may increase in this region with disease progression. To our knowledge, this is the first in vivo
1H-MRS study of bipolar disorder to report such a correlation. Furthermore, none of the
1H-MRS studies of schizophrenia have reported a significant negative correlation between illness duration and hippocampal or medial temporal lobe
N-acetylaspartate measures
(21,
69–77). This finding is consistent with previous findings of 1) a significant negative correlation between right temporal lobe volume and duration of illness in male patients with bipolar disorder
(78), 2) a negative correlation between age at onset of bipolar I disorder and
N-acetylaspartate measures in the right basal ganglia
(47), and 3) negative correlations between illness duration and both right and left prefrontal
N-acetylaspartate measures
(46). Taken together, these findings suggest that illness progression may result in further neuropathological changes in multiple brain regions in bipolar disorder and may perhaps involve brain regions in the right hemisphere more than the left.
With regard to the limitations of the present study, the long-term effects of medications on the hippocampus, which are not known, may have influenced the results. Although no significant associations between
N-acetylaspartate concentrations and either divalproex or lithium doses were found, it is conceivable that long-term exposure to neuroleptics, mood stabilizers, or antidepressants may be associated with alterations in both the structure and neuronal integrity of the hippocampus. Preliminary studies have suggested that both atypical and typical neuroleptics may increase previously low
N-acetylaspartate levels toward more normative values in the anterior cingulate and dorsolateral prefrontal regions in schizophrenia
(79,
80). Studies of healthy volunteers and bipolar disorder patients demonstrated that 4 weeks of lithium administration significantly increased
N-acetylaspartate in the frontal, temporal, parietal, and occipital lobes
(81), as well as significantly increased total gray matter content in the bipolar disorder patients
(81,
82). There is also evidence that valproate activates pathways that regulate BDNF, robustly increases trophic factors such as Bcl-2 and growth cone associated protein, and promotes neurite outgrowth in as well as prolongs survival of human neuroblastoma cells
(20). Although antidepressant effects on
N-acetylaspartate measures have not been investigated, chronic administration of several different classes of antidepressants has been shown to increase neurogenesis in the adult rodent hippocampus
(83–
86), and this effect may be related to antidepressant-induced up-regulation of hippocampal CREB and BDNF
(60–
62). If chronic administration of antidepressants, lithium, and valproate can all potentially increase
N-acetylaspartate by promoting neurogenesis, this would mean that hippocampal
N-acetylaspartate measures in unmedicated bipolar I disorder patients might be even lower than those reported in the present study of mostly medicated patients.
Finally, the number of subjects was small, and alterations in the T1 or T2 of proton metabolites in the hippocampal region of the patients might have contributed to the observed differences. In other words, as a consequence of relaxation time differences, the N-acetylaspartate measures observed in this study may have to some degree reflected differences in metabolite variability as well as metabolite concentrations; however, there is no evidence that N-acetylaspartate in bipolar disorder patients has abnormal relaxation times. Nevertheless, future studies will probably need to determine if there are significant alterations in N-acetylaspartate relaxation times in the hippocampus of patients with bipolar I disorder.