Recent studies have raised the possibility that the aminergic and neurotrophic hypotheses of major depressive disorder are complementary rather than contradictory. Most pertinent for the present study are reports about lower levels of brain-derived neurotrophic factor (BDNF) in depressed patients
(1,
2). These observations are noteworthy, since it has been shown that endogenous BDNF is critical for the normal development and function of central serotonin neurons as well as for the elaboration of behaviors that are mediated by brain serotonergic systems
(3).
We examined the effects of tryptophan depletion and sham depletion on serum BDNF levels in unmedicated patients with remitted major depressive disorder and healthy subjects. We expected that during tryptophan depletion, serum BDNF levels would be lowered in patients with remitted major depressive disorder relative to healthy subjects and also relative to the sham depletion condition.
Method
Subjects were 27 unmedicated patients with remitted major depressive disorder (18 women and nine men; mean age=39.8 years [SD=12.7]; mean baseline Hamilton depression scale score=1.1 [SD=1.2]; mean age at onset=23.8 [SD=8.4]; mean number of previous episodes=3.6 [SD=2.6]) and 20 healthy comparison subjects (11 women and nine men; mean age=33.7 [SD=12.8]; mean baseline Hamilton depression scale score=0.7 [SD=0.8]); all were nonsmokers. Diagnosis was established according to the Structured Clinical Interview for the DSM-IV, nonpatient version. No additional lifetime diagnosis other than major depressive disorder was allowed. Subjects were medically healthy. They were entered into the study after full explanation of the purpose of the study and the study procedures, and after written consent had been obtained as approved by the NIMH Institutional Review Board. The depressed patients had been in remission (Hamilton depression scale total score <8) and had not been taking antidepressant medication for a mean of 40.4 months (SD=48.4) at the time they entered the randomized, placebo-controlled, double-blind crossover study.
Tryptophan depletion was induced by administration of capsules containing 32 g of an amino acid mixture without tryptophan; during sham depletion subjects received identical capsules containing a total of 32 g lactose. Test days were separated by at least 6 days. Serum BDNF levels and plasma tryptophan levels were obtained at baseline and 5, 7, and 24 hours after administration of the capsules on each test day. Behavioral assessments with the Hamilton depression scale were obtained at baseline and 7 and 24 hours after capsule administration.
Analysis of BDNF concentration in the plasma was performed with a ChemiKine BDNF Sandwich ELISA Kit (Chemicon, Temecula, Calif.) according to the manufacturer’s instructions with minor modifications. The samples and standards were applied in duplicate into 96-well immunoplates precoated with rabbit antihuman BDNF antibody and incubated on a shaker overnight at 4°C. After washing, biotinylated mouse anti-BDNF antibody was added and incubated overnight at 4°C. Then streptavidin-HRP was added and incubated at room temperature for 1 hour after washing. TMB substrate was added and incubated at room temperature for 15 minutes. The plate was read with a Multilabel Counter (Wallac, Finland) at 450 nm after added stop solution. The standard curve was linear from 7.8 to 500 pg/ml BDNF. The assay sensitivity is 7.8 pg/ml. The intraassay and interassay variability is within 10%.
For tryptophan level assessments, plasma was deproteinized for total tryptophan or filtered for free tryptophan measurements before it was subjected to an isocratic reversed-phase high-performance liquid chromatography (Waters; excitation/emission wavelengths 300/350 nm).
BDNF data and plasma tryptophan concentrations were analyzed by using repeated measures ANOVA in which time and treatment were the repeated factors and group was the between-subject factor. Significant results were further examined by using Bonferroni-corrected simple effects tests. Results are presented as means and standard deviations.
Results
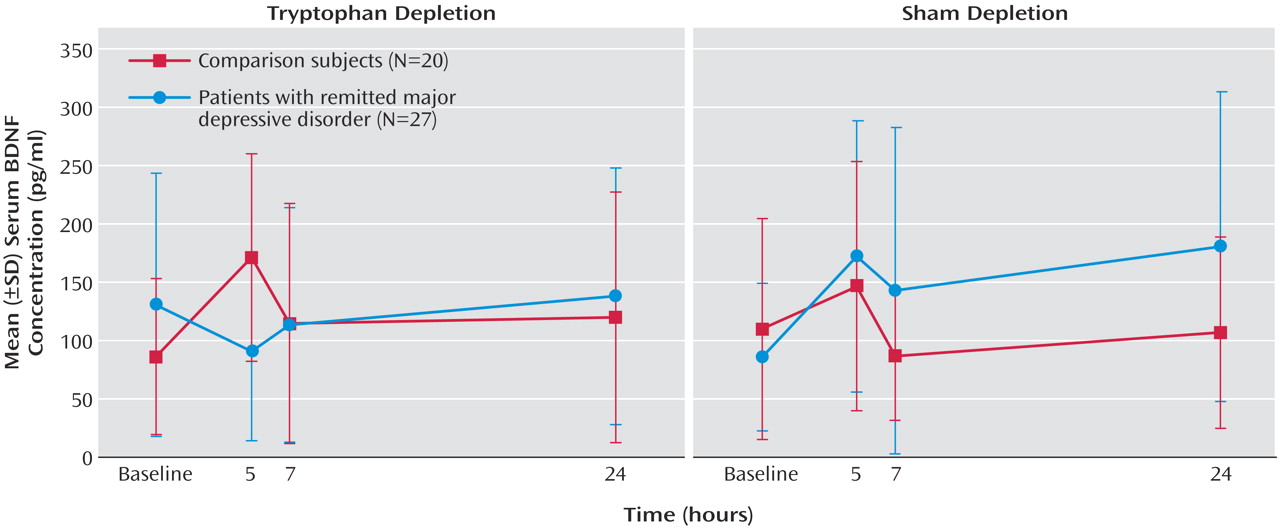
In patients with remitted major depressive disorder, tryptophan depletion but not sham depletion resulted in a transient lowering of BDNF levels at the 5-hour time point. In contrast, tryptophan depletion induced a significant increase in serum BDNF concentrations in healthy subjects. During sham depletion, BDNF concentrations significantly increased in the group with remitted major depressive disorder, but the increase was not significant for the comparison subjects. The treatment-by-time-by-group interaction was significant (F=5.79, df=2.87, 129.13, p<0.001). The time course of changes is described in
Figure 1. No significant correlations were found between Hamilton depression scale scores and BDNF levels at any time point. Changes in BDNF levels did not differ between the 16 remitted major depressive disorder patients who showed a transient return of depressive symptoms during tryptophan depletion (defined as an increase in Hamilton depression scale total score to greater than 10) and the nine patients who remained well. As expected, tryptophan depletion lowered plasma total and free tryptophan levels (treatment-by-time interaction: F=166.75, df=2.25, 96.85, p<0.001, and F=79.99, df=2.79, 125.82, p<0.001) with no between-group differences and nadir values at the 5- and 7-hour time points.
Discussion
During tryptophan depletion, BDNF levels increased in healthy volunteers. By contrast, patients with remitted major depressive disorder did not show a similar response, and BDNF levels remained low in these patients. The transient increase in BDNF concentrations in healthy subjects during tryptophan depletion suggests a compensatory response to maintain the complex interactions between serotonergic and neurotrophic systems. It is interesting that this response appears to be dysfunctional in patients with remitted major depressive disorder, who are unable to mount this compensatory response.
We have previously postulated that antidepressants bring about their beneficial effects via both trophic and neurochemical support
(4); the observations of the present study suggest that the two systems may indeed be intimately linked. Unexpectedly, increases in plasma BDNF were observed during sham depletion in patients with remitted major depressive disorder. This suggests the existence of a complex dysregulation between the systems that may be critical to the vulnerability of depression.
Previous studies have suggested that plasma BDNF may derive from both central and peripheral sources. However, findings that BDNF in the periphery crosses the blood-brain barrier by a high-capacity, saturable transport system
(5), and evidence that serum BDNF levels do not differ from CSF levels, suggest that peripheral changes may also reflect central processes
(6). The time course of changes is particularly relevant because acute changes in plasma BDNF levels in response to tryptophan depletion were measured. In this context, the onset of changes in BDNF mRNA expression in the brain increased as early as 15 minutes after stress exposure.
Baseline BDNF levels did not differ in subjects with remitted major depressive disorder versus healthy subjects. Recent studies have reported lower BDNF levels in depressed patients during a spontaneous episode of major depressive disorder. Our results suggest that it is a dysregulation of BDNF homeostasis in the face of a serotonergic perturbation that truly represents a trait vulnerability marker for depression.