Cerebellar dysfunction has been implicated in the pathophysiology of schizophrenia. For example, Andreasen et al.
(1) suggested that patients with schizophrenia experience cerebellar dysmetria (or ataxia) of thought and that abnormal cerebellar function results in a mismatch between reality and perceived reality, leading to psychotic thinking. In addition, Schmahmann and Sherman
(2) reported that patients with lesions localized to the cerebellum had cognitive deficits similar to those seen in patients with schizophrenia. These include impairments in working memory, abstract reasoning, verbal memory, and executive function. Moreover, smaller vermal volumes
(3) and reduced linear density and size of Purkinje cells in the vermis
(4) have been found in postmortem samples from patients with schizophrenia compared with those from normal comparison subjects. Finally, Nopoulos et al.
(5) suggested that decreased Purkinje inhibitory output results in an increased excitatory drive from the fastigial nucleus to mesencephalic dopaminergic neurons, resulting in a hyperdopaminergic state.
A paradigm to measure cerebellar activity with transcranial magnetic stimulation has been described by Ugawa et al.
(6) and involves the application of a cerebellar conditioning stimulus to the cerebellar cortex 5 to 15 msec before a motor cortex test stimulus, inhibiting the size of the motor evoked potential produced by the test stimulus by approximately 50%
(7). This inhibition, referred to as cerebellar inhibition, is thought to be mediated through transcranial magnetic stimulation activation of inhibitory Purkinje cells, which reduce the excitatory drive from the deep cerebellar nuclei (i.e., dentate and interpositus) to the motor cortex via the ventrolateral nucleus of the thalamus
(6). Therefore, cerebellar inhibition represents an important measure of cerebellar activity and of the integrity of the cerebellar-thalamic-cortical pathway
(8).
The objective of the present study, therefore, was to evaluate cerebellar inhibition in patients with schizophrenia. We hypothesized that patients with schizophrenia would demonstrate deficits in cerebellar inhibition compared with healthy subjects.
Method
The study included 10 right-handed patients (mean age=36.2 years, SD=13.2, nine male and one female) with a DSM-IV diagnosis of either schizophrenia or schizoaffective disorder confirmed by the Structured Clinical Interview for DSM-IV
(9) and 10 right-handed healthy comparison subjects (mean age=30.7 years. SD=8.7, seven male and three female). Of the 10 patients with schizophrenia, three had been free of antipsychotic medication for 1 month or longer and seven were medicated with an atypical antipsychotic (olanzapine: 10, 15, 15, and 20 mg/day, for four subjects, respectively; quetiapine: 100 and 900 mg/day for two subjects, respectively; risperidone: 6 mg/day for one subject). The Centre for Addiction and Mental Health ethics committee approved the study, and written informed consent was obtained from each participant.
Surface electromyography (EMG) was recorded from the right first dorsal interosseous muscles. Subjects maintained relaxation throughout the experiment, the EMG was monitored on a computer screen, and the signal was amplified, filtered (band pass 2 Hz to 2.5 kHz), and digitized at 5 kHz.
Transcranial magnetic stimulation of the left motor cortex was conducted and the motor threshold was determined according to previously published studies
(10) with a 7-cm figure-of-eight coil (The Magstim Company, Whitland, U.K.). The test stimulus was adjusted to produce a motor evoked potential amplitude of about 0.5 mV in the right first dorsal interosseus muscle
(7). The cerebellar conditioning stimulus was performed with a double-cone coil (mean diameter=110 mm) centered over the right cerebellar hemisphere and set at 5% below the threshold for pyramidal tract activation
(7). The cerebellar conditioning stimulus was applied before the test stimulus at one of four random interstimulus intervals (5, 7, 9, and 15 msec). Trials were performed, each consisting of five randomly intermixed conditions presented 10 times each. The time between trials was 5 seconds. Cerebellar inhibition was expressed as a ratio of the conditioned to the unconditioned motor evoked potential amplitude.
Results
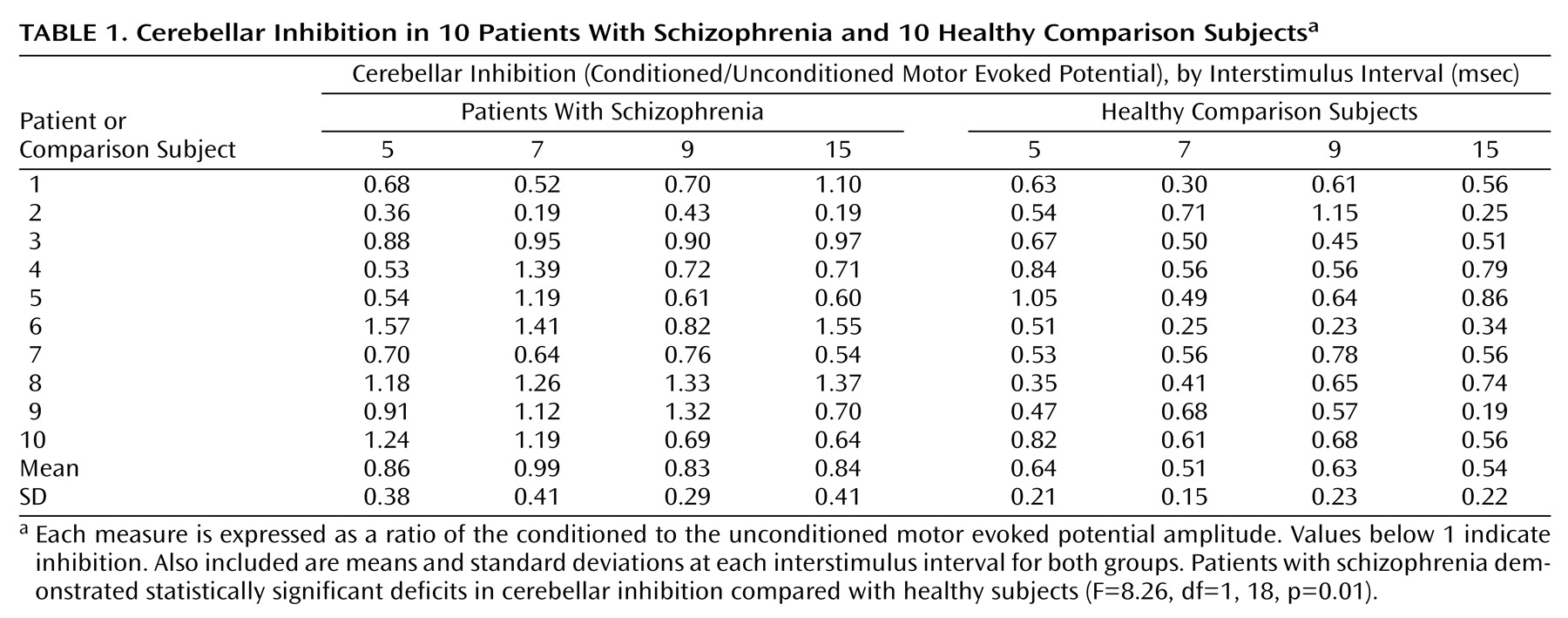
The mean motor evoked potential amplitude for test stimulus alone was 0.56 mV (SD=0.13) for patients with schizophrenia and 0.59 mV (SD=0.16) for healthy comparison subjects and, therefore, closely matched. On measures of inhibition, a significant main effect of group (patients with schizophrenia and healthy comparison subjects) was obtained (
Table 1) (effect size: Cohen’s d=1.02) with no significant interaction of group by interstimulus interval (F=0.02, df=1, 18, p=0.90)
(11). Across all interstimulus intervals, patients with schizophrenia demonstrated 29.8% less inhibition than healthy comparison subjects.
Discussion
Our results demonstrate that patients with schizophrenia have deficits in cerebellar inhibition compared with healthy subjects. These deficits may be the result of either abnormal cerebellar inhibitory output or disrupted cerebellar-cortical connectivity. With regard to the former, inhibitory Purkinje cell output results in a reduction of excitatory output from deep cerebellar nuclei to the cortex, which leads to modification of cortical control
(12). Reyes and Gordon
(13) demonstrated that patients with schizophrenia had a reduction in number of Purkinje cells per unit length of Purkinje cell layer (cells/mm). With regard to the possibility of disrupted cerebellar-cortical connectivity, it has been reported that during recall of novel and practiced word lists in neuroleptic-free patients with schizophrenia there was a reduction in regional cerebral blood flow in the anterior cingulate, thalamus, and cerebellum compared with healthy subjects
(14), suggesting that patients with schizophrenia have altered prefrontal-thalamic-cerebellar connectivity. Therefore, our findings of deficient cerebellar inhibition in patients with schizophrenia provide confirmatory in vivo evidence of either an abnormality in Purkinje cell output or disrupted cerebellar-cortical connectivity.
Cerebellar output may result in changes to cortical interneuron function. In a recent study
(8), we suggested that cerebellar excitation induced by transcranial magnetic stimulation results in a reduction in the excitatory output pathway originating in the deep cerebellar nuclei and terminating on both pyramidal neurons and interneurons in the cortex. Consequently, aberrant cerebellar activity may result in altered inhibitory interneuron activity in the cortex. In view of the fact that altered activity of inhibitory interneurons in the cortex has been posited as a pathophysiological mechanism in schizophrenia
(10,
15), it remains possible that cortical inhibitory dysfunction in schizophrenia may be mediated, in part, through a disturbance in cerebellar activity.
There were several limitations to this study. First, the number of patients and comparison subjects was small; the findings should be replicated with a larger sample. Second, seven patients in this study were receiving a single neuroleptic and only three were unmedicated. Third, although the cerebellar conditioning stimulus activates primarily cerebellar inhibitory mechanisms, it is possible that noncerebellar mechanisms may be involved. For example, Werhahn et al.
(16) demonstrated that at interstimulus intervals of 8 msec or greater, suppression of EMG responses from cortical stimulation may occur, in part, through activation in C6/7 nerve roots in the brachial plexus, resulting in sensory afferent—not cerebellar—inhibition of the motor cortex. Nevertheless, this study provides preliminary in vivo evidence for altered cerebellar inhibitory activity in patients with schizophrenia.