Comparison to Previous In Vivo Imaging Studies
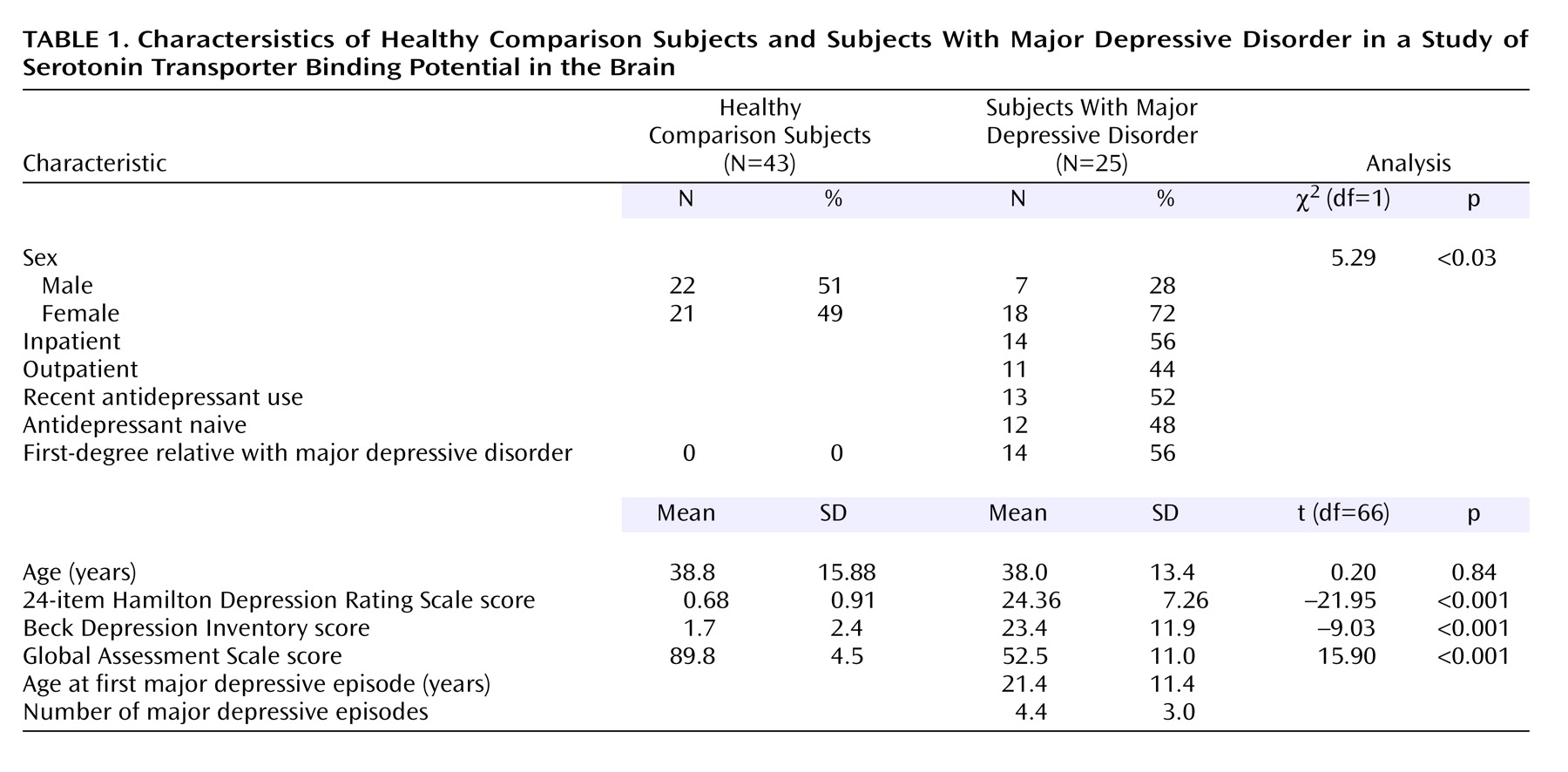
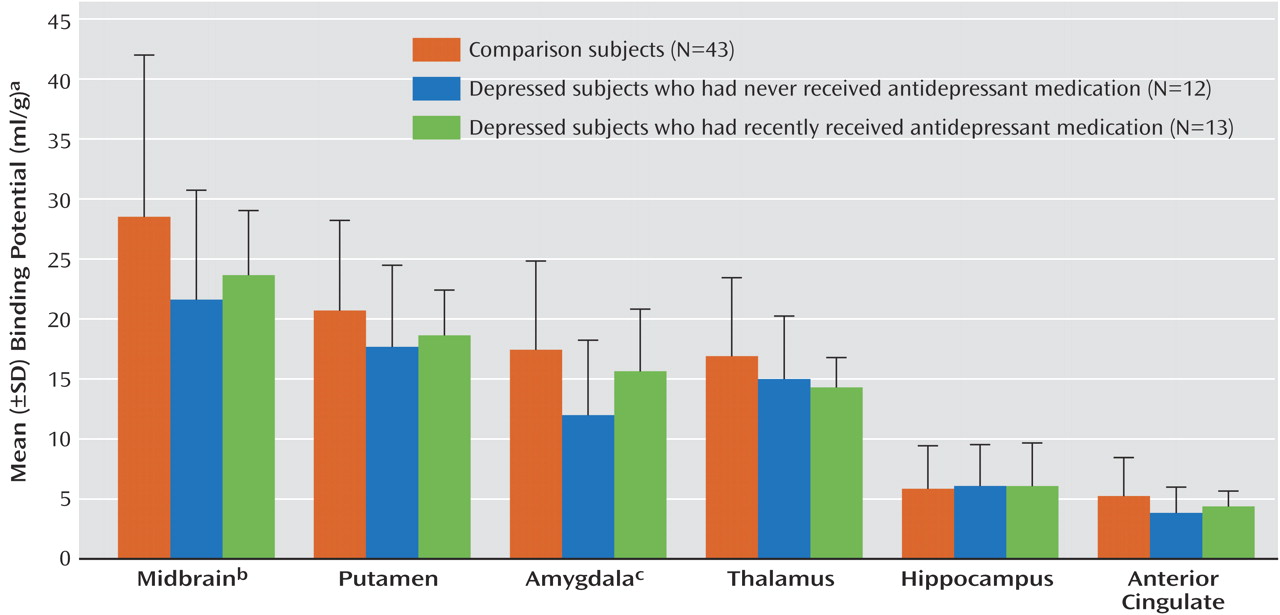
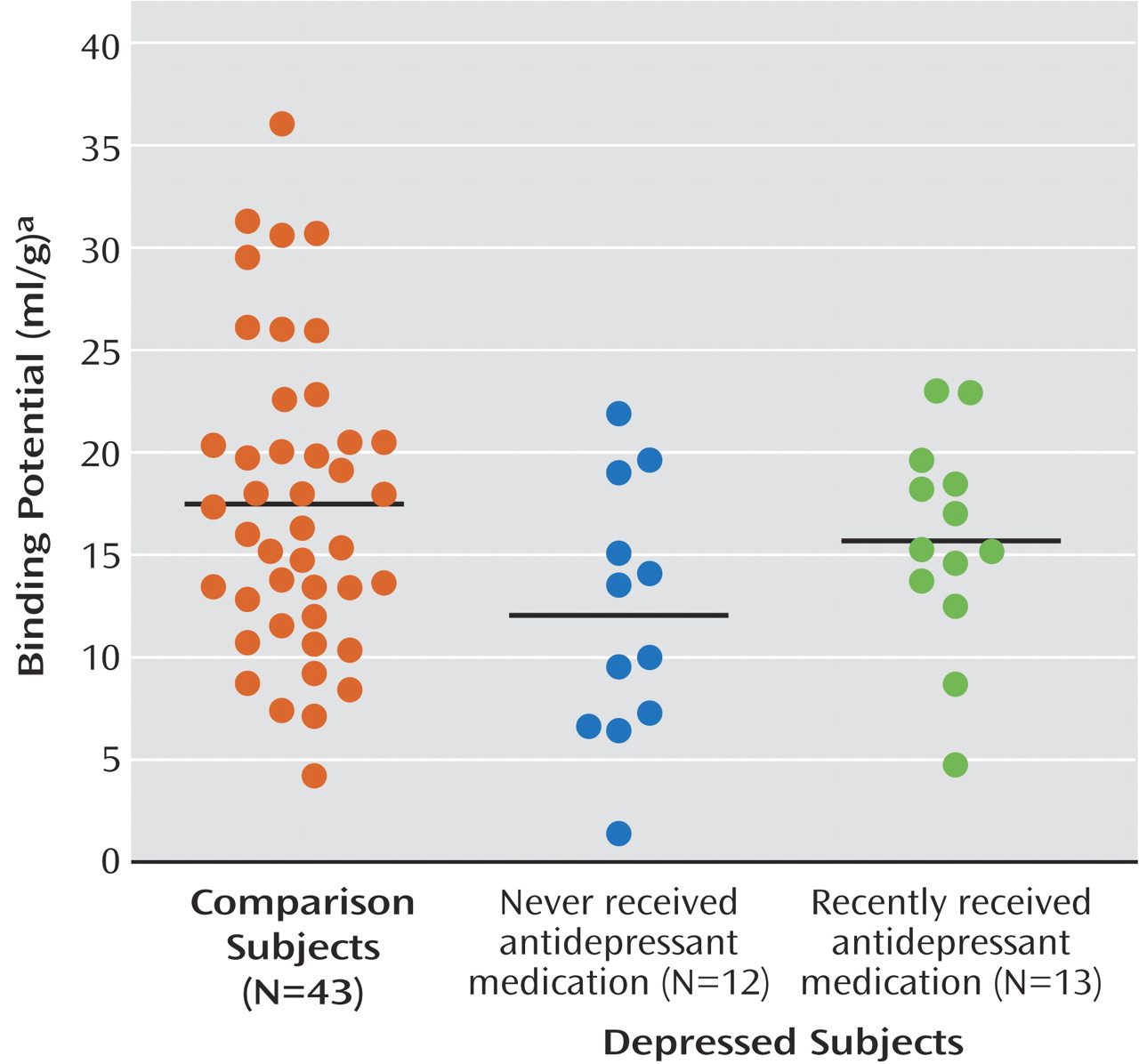
Our finding of a 20% lower BP′ in the midbrain in subjects with major depressive disorder, compared to healthy volunteers, is in agreement with that of Malison et al.
(20), who found a 19% lower [
123I]β-CIT binding in subjects with major depressive disorder, but is at odds with the findings of Ichimiya et al.
(37), who reported no difference in midbrain [
11C]McN 5652 V
3′′ (V
3′′ is defined later in the Discussion section) and
higher V
3′′ in the thalamus of depressed subjects. It should be noted that the study by Ichimiya et al. 1) included euthymic subjects; 2) used data from 90-minute scans; 3) used age as a covariate (we could detect no age-related changes in BP′ in the midbrain); 4) used the graphical method of Logan, which is reported to have noise-dependent bias
(38); and 5) had a smaller number of depressed subjects (N=13). Our data are also consistent with the results of a previous PET study with the transporter radioligand [
11C]
N,N-dimethyl-2-(2-amino-4-cyanophenylthio)benzylamine ([
11C]DASB) in which no difference in striatal V
3′′ was found between depressed subjects and healthy comparison subjects
(39). We found a 20% lower BP′ in the amygdala in subjects with major depressive disorder, compared to healthy subjects; to our knowledge, this finding has not been reported previously.
Interpretation of Findings
We interpret the lower [
11C]McN 5652 BP′ (f
1B
max/K
d) in the subjects with major depressive disorder to reflect lower B
max, or lower total number of available serotonin transporter binding sites. Several outcome measures are used in PET studies, including BP (B
max/K
d=V
T – V
2/f
1), BP′ (f
1B
max/K
d=V
T – V
2), and V
3′′ (f
2B
max/K
d=BP′/V
2), where
VT is the total volume of distribution in an region of interest,
V2 is a measure of the free and nonspecifically bound tracer,
f1 is the free concentration of the radiotracer in plasma, and
f2 is the free concentration of the radiotracer in the cerebrospinal fluid. Ideally we would measure BP, but f
1 is not measurable with [
11C]McN 5652. Our findings were similar when either of the outcome measures that can be determined with [
11C]McN 5652 was used, i.e., the principal region-by-diagnosis interaction was significant with BP′ or V
3′′ (p<0.05), and the interaction of region and prior antidepressant medication status was significant with V
3′′ (p<0.03). We believe the lower BP′ in major depressive disorder is not a consequence of f
1, because our findings are consistent with postmortem findings, where f
1 is not a factor, and because changes in f
1 would result in global differences, not regionally specific ones. Finally, postmortem studies suggested that lower levels of serotonin transporter in subjects with major depressive disorder are not due to changes in receptor affinity (1/
d)
(14). It is unlikely that the lower BP′ in major depressive disorder could be a consequence of
higher synaptic levels of 5-HT, because of the hypothesized serotonin abnormality in depression. Moreover, in postmortem studies that included a preincubation period to promote catabolism and a washout of endogenous 5-HT before adding ligand in vitro, lower serotonin transporter binding was found in major depressive disorder
(40–
42). These findings appear to rule out elevated intrasynaptic levels of 5-HT as an explanation. Therefore, our finding of lower serotonin transporter BP′ in major depressive disorder is likely a result of lower serotonin transporter B
max, i.e., fewer serotonin transporters.
There are several possible explanations for the lower serotonin transporter B
max in major depressive disorder. Lower levels of synaptic 5-HT could facilitate serotonin transporter internalization
(6); there could be fewer 5-HT neurons or neuronal processes or decreased expression of serotonin transporter per terminal; or a combination of these factors could be operating. Lower levels of synaptic 5-HT in the raphe nuclei could be related to the presence of the C(-1019)G polymorphism of the promoter region of the 5-HT
1A gene that has been shown to be overrepresented in patients with major depressive disorder
(43). The C(-1019) allele is part of a 26-base pair imperfect palindrome with less affinity for the nuclear deformed epidermal autoregulatory factor transcriptional repressor, and this lower level of affinity could potentially result in a higher level of expression of the 5-HT
1A protein in the raphe nuclei. Because the raphe nuclei 5-HT
1A receptors are inhibitory, higher levels of 5-HT
1A in the raphe nuclei would result in lower levels of 5-HT release and possible down-regulation of serotonin transporter. Alternatively, lower levels of synaptic 5-HT could be related to having a variant of the tryptophan hydroxylase-2 gene, which has less catalytic activity and is present in 10% of subjects with major depressive disorder
(44,
45). Down-regulation of serotonin transporter should be accompanied by reduced mRNA for serotonin transporter. Our previous autoradiography studies suggested that the number of neurons that express serotonin transporter mRNA is reduced by 54% in subjects with major depressive disorder who commit suicide
(42), although the number of 5-HT-producing neurons did not appear to be decreased
(46).
We hypothesized that a lower level of serotonin transporter in all six regions of interest was necessary for the expression of major depressive disorder, but we found differences in serotonin transporter binding potential only in the midbrain and amygdala. These findings can be interpreted in several ways. First, it is possible that the midbrain and amygdala are the crucial structures in the pathophysiology of depression, and serotonin transporter abnormalities in these regions are sufficient to result in major depressive disorder. Second, the abnormalities in the other regions of interest may not be specific to serotonin transporter but perhaps may involve other measures of serotonergic function. Third, the lower level of serotonin transporter BP′ in the midbrain and amygdala may be secondary to as yet undetermined primary pathological process(es). Finally, the PET ligand we used may not be sensitive enough to detect serotonin transporter abnormalities in the other regions.
For the differences in serotonin transporter BP′ to be limited to the midbrain and amygdala, the abnormality may be confined to a subpopulation of serotonin-transporter-expressing 5-HT neurons within the dorsal raphe nuclei. Less 5-HT input to the amygdala, as suggested by the finding of lower serotonin transporter BP′, may result in increased amygdala activity
(47), as serotonin enhances inhibition in the amygdala, presumably through activation of γ-aminobutyric-acid-ergic interneurons
(48). Serotonergic projections to the amygdala are implicated in anxiety disorders as well
(49). Previous research has suggested an association between hyperactivity in the amygdala and a greater likelihood that sensory or social stimuli are perceived or remembered as emotionally arousing or aversive
(50); thus, hyperactivity in the amygdala may contribute to the relationship between adverse childhood experiences and adult mood disorders.