After 50 years of experience with antidepressant drugs, the large individual variation in antidepressant treatment outcome remains poorly understood. Investigations of biologically related individuals point to a genetic contribution to treatment responsiveness (
1), and several polymorphisms in candidate genes have been associated with antidepressant response (
2,
3). However, few pharmacogenetic associations have been replicated, and they explain only a small fraction of individual differences in antidepressant response. Since our understanding of the mechanisms of action of antidepressants is incomplete, important genes and regulatory intergenic sequences may have been missed in candidate gene studies. Therefore, a systematic exploration of variation across the genome has the potential to detect further variants that may help in understanding the biology of antidepressant action and predict response to a specific antidepressant in an individual patient. In the present study, we report on a genome-wide pharmacogenetic analysis of more than 500,000 common genetic variants, tested for association with change in depression severity over a 12-week period after commencing treatment with a serotonergic or noradrenergic antidepressant among subjects with moderate to severe depression. We conducted four primary analyses to address general and drug-specific effects of genes on antidepressant response and gene-drug interactions. We used secondary analyses to explore the influence of variants in 72 candidate genes on antidepressant action.
Discussion
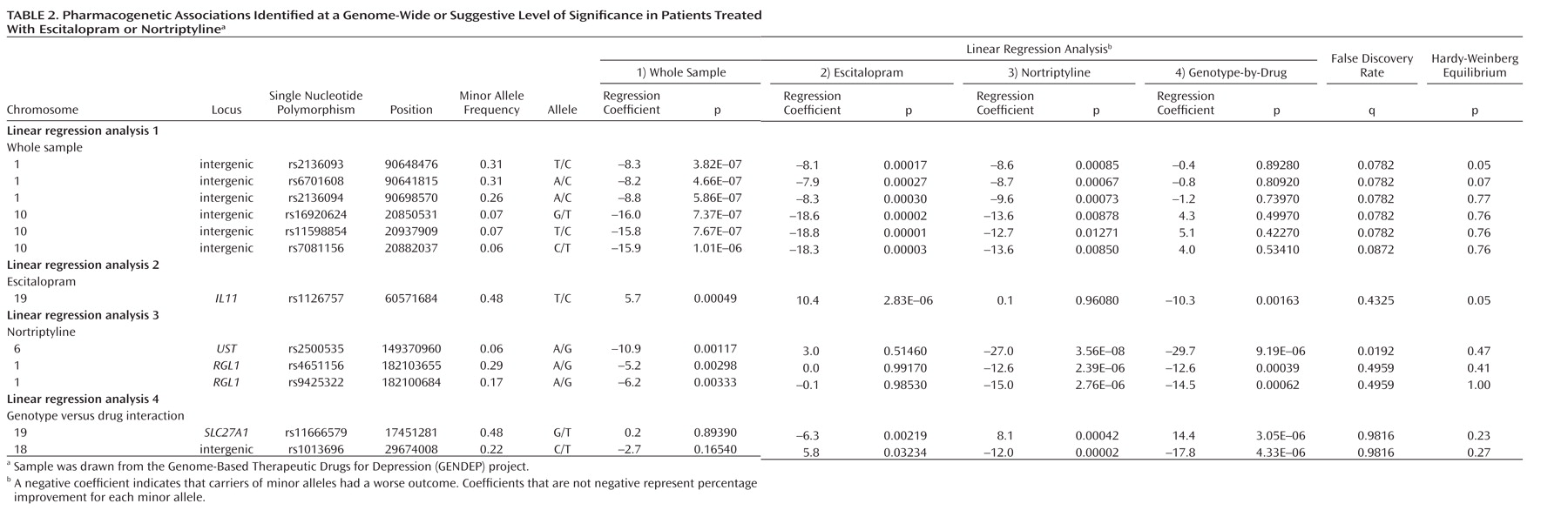
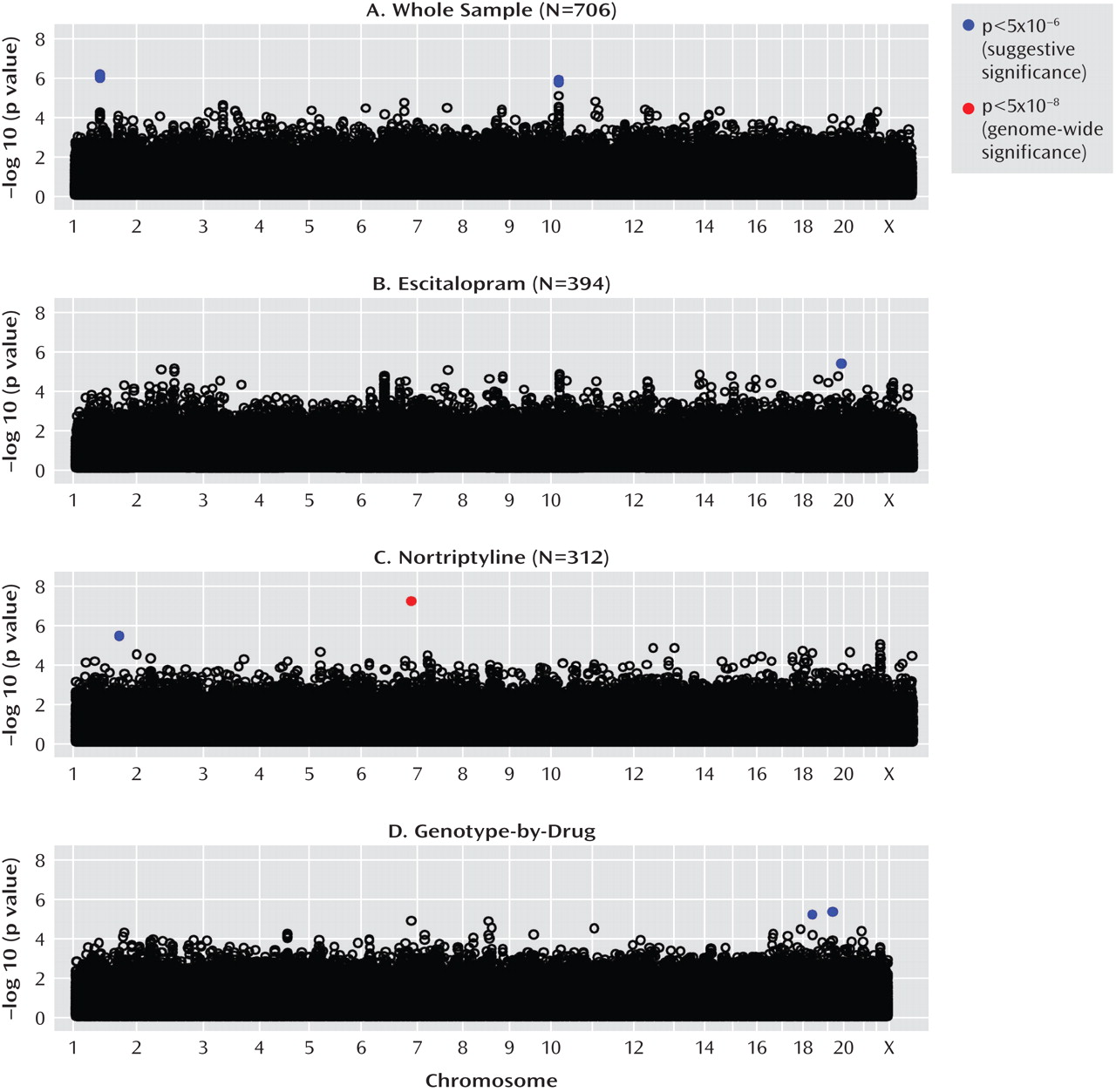
This genome-wide analysis of the GENDEP project has shown that previously unexpected genomic regions may be more potent predictors of antidepressant response than functional candidate genes. Several genomic loci emerged that were more likely to be associated with response than not, after taking into account the extensive multiple testing performed. The genes encoding uronyl 2-sulphotransferase and IL11 as well as two intergenic regions containing common structural variants were revealed as potentially important for antidepressant action. To our knowledge, none of the markers associated with response in the GENDEP project has previously been reported in genome-wide studies of depression susceptibility (
26,
27) or antidepressant response (
28). Since the statistical power of the present investigation was limited, it is likely that a number of moderately strong associations have been missed, and integration of data from multiple large samples may reveal further important pharmacogenetic associations.
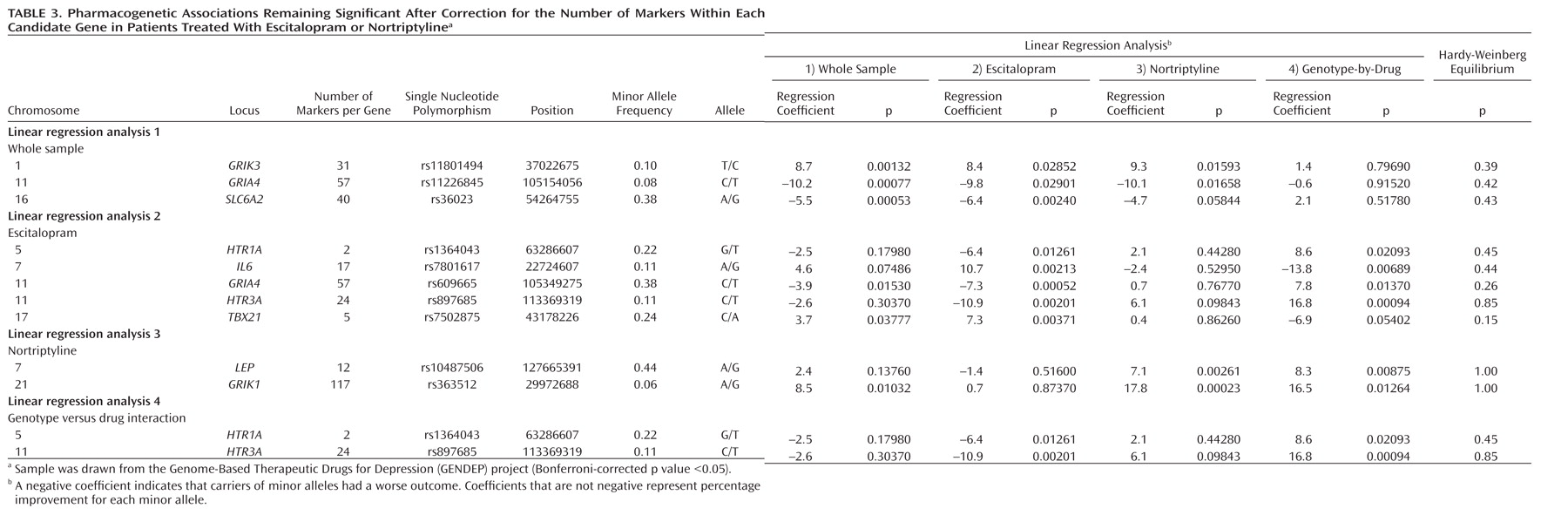
Irrespective of which antidepressant was used, the outcome of treatment was associated with polymorphisms in two regions on chromosomes 1 and 10. The nearest known gene to the associated locus on chromosome 1 (zinc finger protein 326 [ZNF326]) was 380 kilo base pairs away from the associated markers, and the nearest gene to the locus on chromosome 10 (plexin domain containing 2 [PLXDC2]) was 241 kilo base pairs away from the associated markers. Common copy number polymorphisms that may influence the expression of genes at longer distances through structural changes of the chromatin have been reported in both regions. This finding demonstrates the importance of exploring the whole genome rather than concentrating on coding regions of known genes and their flanking sequences.
Antidepressant-specific analyses revealed a strong genome-wide significant association between a marker in the
UST gene and response to nortriptyline. Although the
UST gene did not appear on any previous list of candidate genes, the function of uronyl 2-sulphotransferase in the nervous system suggests a mechanism for its involvement in antidepressant action. This enzyme is responsible for the production of oversulfated proteoglycans that are essential for neurogenesis and neuronal migration (
29). A knockdown of
UST led to severe disturbance of neuronal migration that was rescued by a
UST cDNA (
30). The apparent delayed onset of the pharmacogenetic effect on response after 4 weeks of treatment was consistent with a neurogenesis-related mechanism. It is presently unclear why such an effect should be specific to nortriptyline, since various types of antidepressants increase neurogenesis (
31). Response to nortriptyline was also associated with markers in the
RGL1 gene, involved in Ras and Ral signaling pathways of neuronal differentiation and outgrowth (
32). Nearby markers in this gene have been associated with conduct disorder and hyperactivity (
33). These findings require replication and investigation of the role of neurogenesis-related genes in antidepressant action.
Escitalopram response was predicted by a marker in the gene encoding interleukin-11. Although statistically less robust, this finding was further supported by an association of escitalopram response with a marker in the
IL6 gene, an established candidate gene for depression (
34) and a close homologue of
IL11. Interleukins 6 and 11 are members of a family of neuropoietic cytokines, which are widely expressed in the brain (
35) and can potently inhibit serotonin signaling by inducing raphe neurons to produce acetylcholine instead of serotonin (
36). A differential expression of these cytokines, or alternative splicing suggested by an exonic splicing enhancer consensus sequence (
25), may make some individuals vulnerable to a subtype of depression that may be related to environmental exposures (
37) and poor response to serotonergic antidepressants (
38). This molecular mechanism is consistent with the role of inflammation in a subtype of depression (
39) and could explain the specific moderation by
IL6 and
IL11 of response to the serotonergic antidepressant escitalopram.
A comprehensive list of 72 candidate genes, including those encoding key proteins in monoaminergic and glutamatergic signaling, have produced findings consistent with chance. However, markers in several candidate genes, including
IL6,
HTR3A, and
GRIA4, showed gene-wide significant pharmacogenetic associations that would have been reported as positive results in a candidate gene study. The implication of
IL6 in response to serotonergic antidepressants was supported by the pharmacogenetic association with a functionally related, but genomically independent,
IL11 gene. The
HTR3A gene, encoding the serotonergic 5-HT
3A receptor, is an important candidate as effective antidepressants are antagonists of this receptor (
40) and colocalize with the receptor on cell surfaces (
41). These genes should be considered for exploration in future studies.
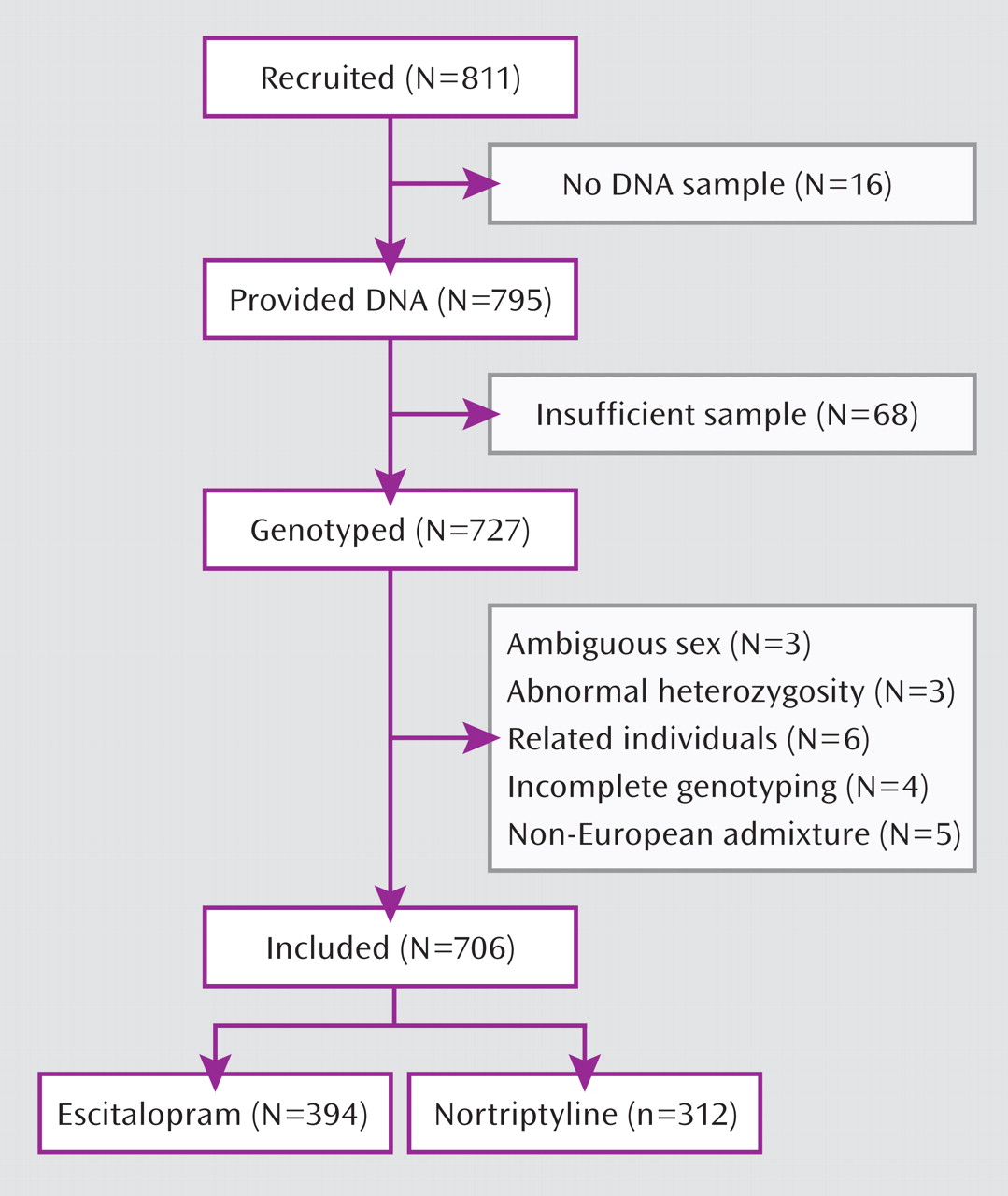
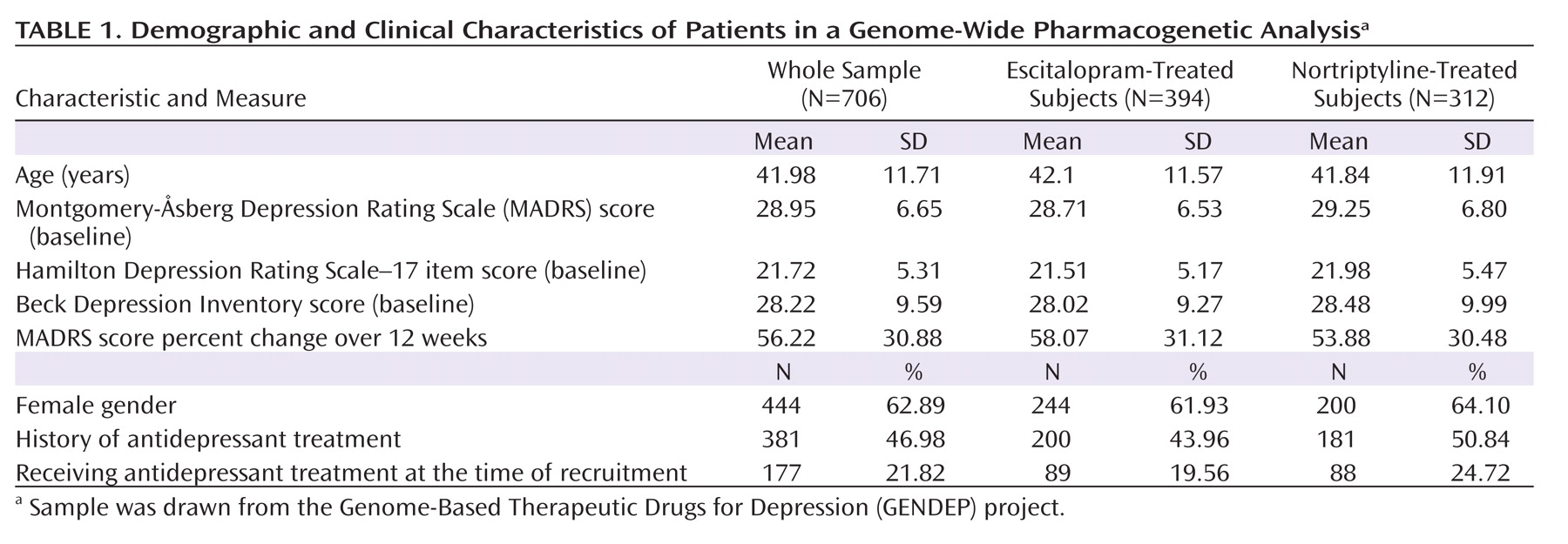
A genome-wide pharmacogenetic study faces a number of challenges. The strengths of the GENDEP project include a homogenous sample specifically recruited for a pharmacogenetic investigation, detailed and complete data characterizing response to antidepressants, tight control of population stratification with no overall inflation of test statistics, and high-quality genotyping enabling stringent quality control while retaining most genotyped individuals. On the other hand, our findings are limited by the absence of a placebo-comparison group, which makes it impossible to distinguish between genetic predictors of nonspecific improvement and effects that are common to both antidepressants used in the GENDEP project. Across the four analyses, only one pharmacogenetic association was established at a genome-wide level of significance. The predominance of negative findings could be the result of limited power to detect weak to moderate associations or of the absence of even moderately strong common genetic determinants of antidepressant drug response. A meta-analysis of multiple large samples will be needed to establish the generalizability of findings reported in the present study.
The pharmacogenetic analysis of GENDEP has brought up a number of promising results, including two regions containing structural variants, a genome-wide significant association with nortriptyline response, and a functionally plausible marker associated with response to escitalopram. These results demonstrate the feasibility of genome-wide pharmacogenetic analyses with well-characterized moderately large samples. As in all genetic studies, replications will be required before the value of any particular finding can be fully assessed, but genome-wide studies may thus be important steps in the elucidation of the genetic basis of pharmacological response.
Acknowledgments
The authors thank the following individuals: Maja Bajs, Mara Barreto, Cristian Bonvicini, Desmond Campbell, Elzbieta Cegielska, Monika Dmitrzak-Weglarz, Amanda Elkin, Caterina Giovannini, Joanna M. Gray, Cerisse Gunasinghe, Bhanu Gupta, Susanne Höfels, Petra Kalember, Pawel Kapelski, Zrnka Kovacic, Dejan Kozel, Anna Leszczynska-Rodziewicz, Sylvie Linotte, Andrej Marusic, Julien Mendlewicz, Metka Paragi, Laura Pedrini, Jorge Perez, Ute Pfeiffer, Aleksandra Rajewska-Rager, Luciana Rillosi, Christine Schmäl, Anna Schuhmacher, Maria Skibinska, Rebecca Smith, Anne Schinkel Stamp, Jana Strohmaier, Jerneja Sveticic, Aleksandra Szczepankiewicz, Alenka Tancic, Sandra Weber, and Richard J. Williamson.