In this issue of
The American Journal of Psychiatry, Burdick et al. (
1) examine the
MET proto-oncogene (
MET) as a candidate gene for schizophrenia in an effort to uncover a potential molecular basis for the inverse relationship between schizophrenia and cancer. The authors studied 16 nonredundant single nucleotide polymorphisms (SNPs), genotyped as part of a genome-wide association (GWA) marker panel, that formed four primary haplotype blocks accounting for 71% of the common allelic variation in
MET. In the initial sample, consisting of 173 schizophrenia cases and 137 comparison subjects, the common allele of one of these haplotype blocks was found to be overexpressed among comparison subjects (47%) relative to patients (33%). This result was replicated in a sample consisting of 107 cases and 112 comparison subjects, where again comparison subjects overexpressed the common haplotype (46%) relative to patients (36%). Several SNPs within this haplotype block were found to show a similar pattern of association with schizophrenia, independently in the initial and replication samples. In a secondary analysis, the authors found that the allele overexpressed in comparison subjects was also associated with increased cognitive ability, defined as higher scores on the first principal component of multiple neurocognitive test indices.
If
MET is truly related to susceptibility for schizophrenia, it is reasonable to wonder why this locus has not emerged from recent large-scale GWA investigations (
2,
3). In fact, very few loci (and none of the major candidate genes such as DISC1, dysbindin, and neuregulin) have emerged as significantly associated with schizophrenia in the extant GWA studies, a pattern that is likely to reflect at least in part the complexity of the inheritance of the disorder, which involves many genes, each with a very small effect on disease risk (4). Because GWA studies examine hundreds of thousands to millions of SNPs for association with diagnosis simultaneously, they must incorporate a substantial correction for multiple hypothesis testing, which results in loss of statistical power to detect genes of small effect. Despite their comparatively small sample sizes, candidate gene studies such as this one examine only a small number of polymorphisms and consequently may have relatively greater statistical power than GWA studies, but the results must meet rigorous tests for both biological plausibility and statistical validity.
There are at least three aspects of biological plausibility that merit discussion in this context. The first is the epidemiological finding of lower than expected rates of cancer in schizophrenia patients and their relatives despite increased prevalence of cancer risk factors such as smoking and obesity (
5,
6). This finding, together with the absence of evidence implicating any nongenetic mechanism in this association, lends credence to the hypothesis that inherited factors may underlie the inverse epidemiological relationship between schizophrenia and cancer.
The nature of the observed molecular association of
MET with schizophrenia is suggestive of antagonistic plieotropy. That is, the allele overtransmitted to healthy subjects is associated with opposite effects on two biomedically negative outcomes—increased cancer risk (probably through increased
MET expression) but decreased schizophrenia risk. Although there are few known examples of antagonistic plieotropy in biomedicine (cf., sickle-cell carriers and protection against malaria), such effects could help to explain the persistence of some schizophrenia-related genetic variants in the population despite reduced fecundity (
7). If the
MET-schizophrenia association operates in this way, one would expect over-transmission of cancer-
protecting variants to patients with schizophrenia, a hypothesis that is likely to require a much larger sample size to test with adequate power.
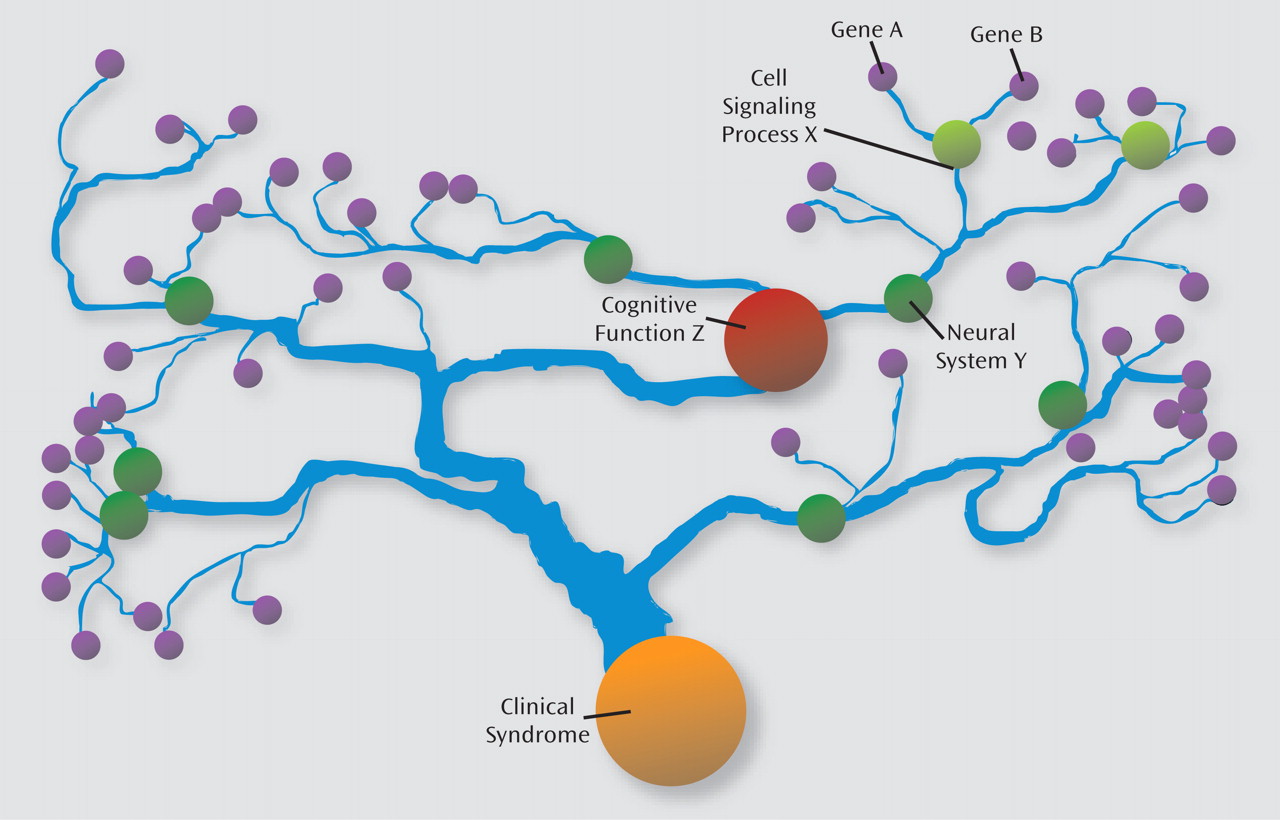
In the genetics of complex neuropsychiatric syndromes such as schizophrenia, individual gene effects on the illness phenotype are expected to be smaller than those on intermediate phenotypes that are theoretically closer to the mechanism of gene action (
1). Such complexity may be analogous to a watershed model (
Figure 1). Although each gene has a particular set of impacts on cellular signaling, the effects of multiple genes "flow together" in influencing the structure and function of large groups of neurons, thereby affecting neural system activity and dimensions of cognition and behavior impacted in the illness. Genetic modeling studies have documented a substantial genetic correlation between schizophrenia and lower intellectual functioning (
9). That the
MET allele overtransmitted to healthy individuals and undertransmitted to patients with schizophrenia was also associated with higher intellectual functioning is consistent both with the endophenotype-watershed model of schizophrenia inheritance and the substantial genetic covariance between schizophrenia and lower intellectual functioning. Note, however, that this
MET variant accounted for only about 1% of general cognitive ability; therefore, presumably its effect on schizophrenia risk is at least this small and likely much smaller. In addition, it is puzzling why the
MET-intelligence relationship was statistically significant in the combined sample (initial and replication) of comparison subjects, but not in the combined sample of patients (although trending in the same direction), despite comparable sample sizes.
Candidate gene studies also carry a substantial burden to prove that the genetic differences between patients and healthy subjects are not an artifact of population stratification (i.e., chance admixture of different ancestral populations) and or multiple testing (i.e., type I statistical error). The authors addressed the population stratification issue in two ways. First, they performed a two-stage analysis, with replication in an independent sample of schizophrenia patients and comparison subjects. This result reduces the likelihood of stratification effects given that the same pattern of chance genetic admixture is unlikely in two independently ascertained samples. Second, the authors performed a principle components analysis of the GWA marker panel and covaried for the two components found to differ between patients and healthy subjects when testing for the associations of schizophrenia with
MET polymorphisms. If the
MET association with schizophrenia in fact results from chance admixture of two ancestral populations, this analysis of covariance procedure would be expected to nullify it (
10).
Nevertheless, every study includes some degree of sampling error, and it is therefore critical to consider whether the differences in allele frequencies between schizophrenia patients and comparison subjects are due to deviations from expected allele frequencies in the healthy subjects, rather than in the patients. For some of the SNPs in the associating haplotype block, the allele frequencies observed in schizophrenia cases appear identical to those listed in HapMap for European Caucasians. In contrast, the allele frequencies observed in the comparison group appear somewhat lower than those in the HapMap database. The HapMap CEU population is derived from a small geographic region in Utah; given that the schizophrenia cohort in this study was collected in Eastern Queens/Long Island, N.Y., the gene frequencies for healthy subjects (who were further screened for absence of psychiatric morbidity in relatives) drawn from the same geographic/demographic pool would nevertheless appear to provide a better basis of comparison.
The authors addressed the multiple comparisons issue in two ways, first by computing Bonferroni adjustments of the significance criterion (alpha) according to the number of haplotypes examined and second by performing permutation analysis of the association of schizophrenia with allelic variation in the associating haplotype block. Of course, the requirement of significant association across two independent samples of schizophrenia patients and comparison subjects also speaks to the issue of statistical validity. Although some argue that candidate gene studies must still meet statistical criteria for genome-wide significance, such a conservative threshold seems overly stringent, particularly in the context of a disorder with no (known) major gene effects.
Overall, this article by Burdick et al. demonstrates that with careful analysis of biological plausibility and application of rigorous measures to protect against statistical and admixture artifacts, candidate gene studies continue to have a role in the GWA era of psychiatric genetics.