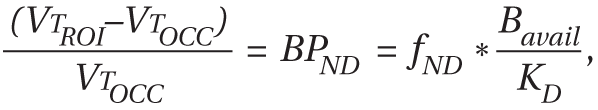Positron emission tomography (PET) imaging studies have demonstrated lower baseline dopamine levels and less induction of dopamine release by stimulants (methylphenidate or
d-amphetamine) in the striatum of chronic cocaine abusers relative to matched healthy comparison subjects (
1–
4). These studies also suggest that lower dopamine release in the ventral striatum in abstinent cocaine abusers is associated with relapse (
2,
4). A possible mechanism leading to lower stimulant-induced dopamine release in cocaine abusers may be related to the loss of dopaminergic terminals from chronic cocaine abuse. As the dopamine transporter is located exclusively in dopaminergic terminals (
5), previous studies focused on the differences in dopamine transporter availability to measure dopaminergic terminals in cocaine abusers and comparison subjects. Unfortunately, the results of these postmortem and imaging studies evaluating the dopamine transporter in chronic cocaine abusers were mixed and inconclusive (
6). It is likely that the variation in the time since last use of cocaine (i.e., abstinence period) in different patient cohorts led to these discrepant findings (
6). This issue has posed a major obstacle to reliable assessment of the available dopamine transporter in cocaine abusers because the dopamine transporter is highly vulnerable to regulation by drugs such as cocaine that alter synaptic dopamine concentrations (
7,
8). This led to the investigation of other markers of dopaminergic terminals, such as the vesicular monoamine transporter, type 2 (VMAT2), in cocaine abusers.
VMAT2 is active in presynaptic vesicular membranes (
9) and is involved in the transport of the various monoamines, such as serotonin, norepinephrine, and dopamine, from the cytoplasm to their storage vesicles. While VMAT2 is not specific to the vesicle of one particular monoaminergic terminal, VMAT2 in the striatum has been reported to largely (>95%) represent storage vesicles in the dopaminergic terminals (
10). Four postmortem studies have contrasted the VMAT2 density in cocaine abusers and matched healthy comparison subjects with radiolabeled derivatives of dihydrotetrabenazine (DTBZ), as they are selective and specific for VMAT2 (
11). Three out of these four studies showed significantly less [
3H]DTBZ binding (range: –12% to –22%) in the striatum of cocaine abusers relative to comparison subjects (
12–
15). As lower VMAT2 binding in cocaine abusers was associated with higher dopamine transporter binding in the study by Little and colleagues (
13), the authors hypothesized that individuals who had experienced greater exposure to cocaine (as evidenced by a compensatory up-regulation of dopamine transporter) lost more dopamine terminals (as evidenced by lower VMAT2) because of toxicity. Lower VMAT2 binding in cocaine abusers, which results from a reduction in the number of dopamine storage vesicles and/or loss of presynaptic dopamine terminals, is important because it informs the mechanisms that lead to the blunting of stimulant-induced dopamine release in cocaine dependence. As we know of no in vivo studies that have confirmed the lower VMAT2 binding reported in the in vitro literature, we studied the impact of chronic cocaine exposure on striatal VMAT2 nondisplaceable binding potential (BP
ND) using PET and [
11C]DTBZ in a group of 12 chronic cocaine abusers and 12 healthy comparison subjects matched for age, gender, race, and nicotine smoking status.
Table 1 contains a glossary of terms relevant to this study.
Results
Twelve cocaine-dependent subjects (mean age=43 years, SD=8) and 12 healthy comparison subjects (mean age=41 years, SD=8) were enrolled in this study; each group contained four women and eight men. The groups were matched as well as possible on both ethnicity (cocaine abusers: eight African Americans and four Caucasians; comparison subjects: five African Americans and seven Caucasians) and smoking status (seven smokers in each group). The cocaine abusers reported smoking crack cocaine for a mean of 18 years (SD=7) and were spending an average of $560 (SD=$480) weekly.
Scan Variables
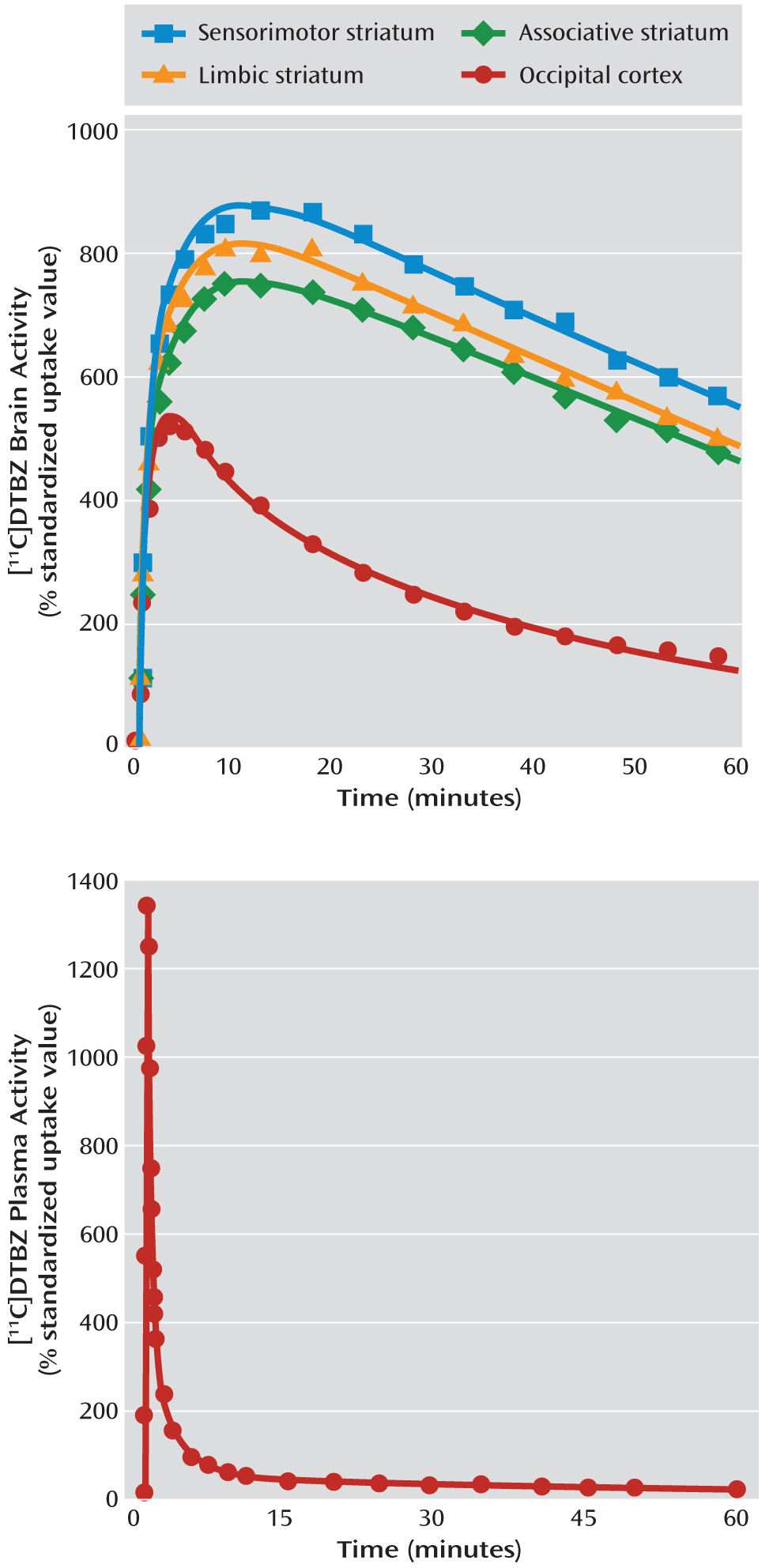
Critical PET scan variables are listed in
Table 2. The [
11C]DTBZ injected dose, specific activity at the time of injection, and injected mass did not differ between the groups. No significant between-group differences were observed in the rate of [
11C]DTBZ clearance from the plasma compartment or in the [
11C]DTBZ occipital cortex distribution volume, V
ND (data available from the 10 subjects in each group for whom arterial line placement was successful).
Regional Volumes
No significant between-group differences were found in the volumes of the regions of interest or reference region (
Table 3), suggesting a lack of measurable volumetric changes in the human striatum after chronic cocaine abuse.
Measurement of VMAT2 Availability
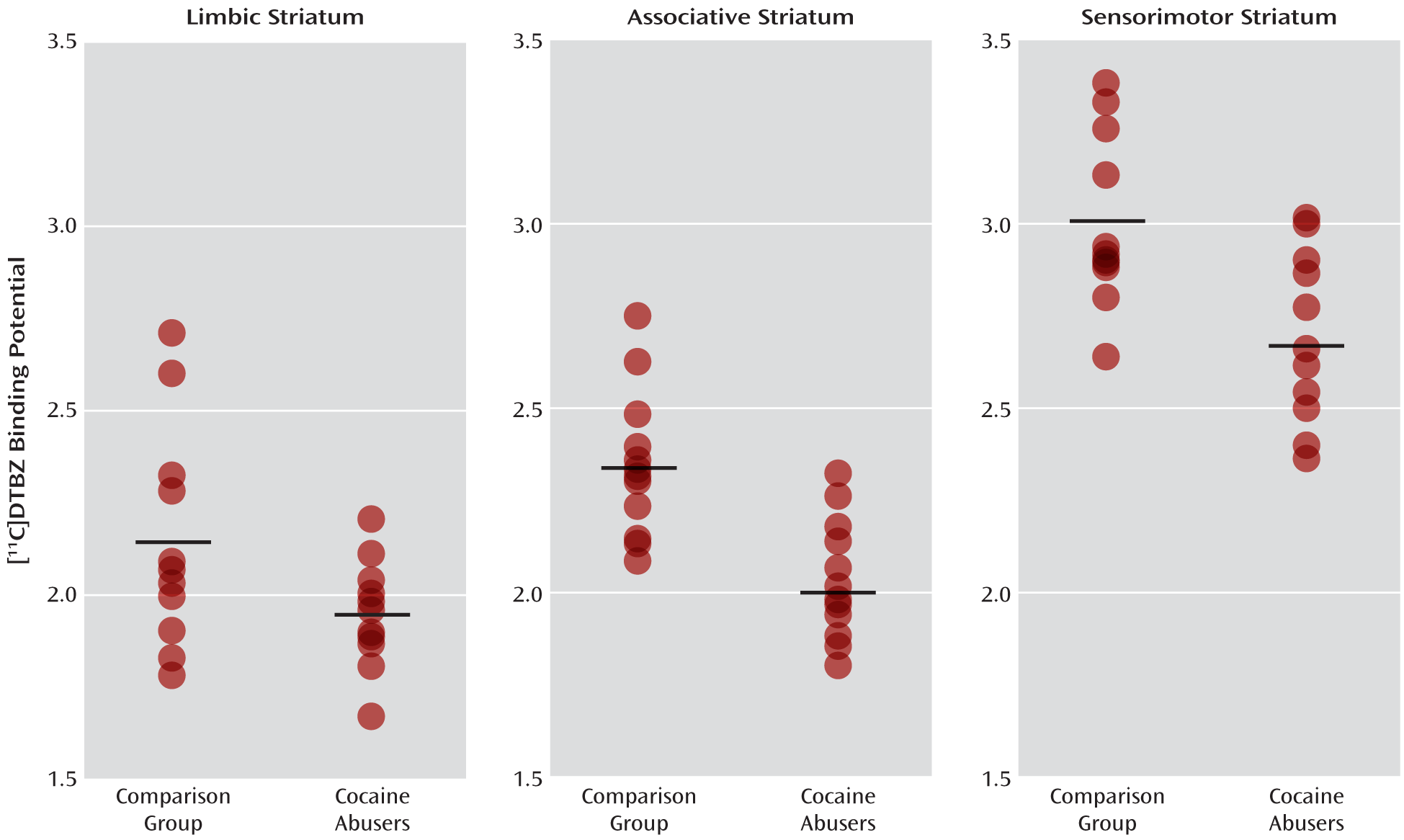
The MANOVA performed on BPND data derived with kinetic analysis for 10 subjects in each group and with the simplified reference tissue method for two subjects per group demonstrated that the cocaine abusers had significantly lower [11C]DTBZ BPND in the striatal subdivisions relative to the healthy comparison subjects (effect of diagnosis: F=4.98, df=5, 18, p=0.005).
The MANOVA performed on BPND data derived with the simplified reference tissue method for all of the subjects in both groups also demonstrated that the cocaine abusers had significantly lower [11C]DTBZ BPND in the striatal subdivisions than the comparison subjects (effect of diagnosis: F=4.52, df=5, 18, p=0.008).
Values for [
11C]DTBZ BP
ND in the striatum and its subdivisions derived by using a combination of kinetic analysis and the simplified reference tissue method are shown in
Table 4; those derived by using only the simplified reference tissue method are shown in
Table 5. Also included in these tables are the results of the unpaired t tests that were used to contrast the individual striatal subdivisions. The p values of the five anatomical subdivisions of the striatum in
Tables 4 and
5 remained significant (<0.05) after we applied the false discovery rate correction for multiple comparisons. Individual values for the three functional striatal subdivisions appear in
Figure 2.
Age-corrected correlation analyses revealed no significant associations between VMAT2 availability in the striatum and the number of years of cocaine use (r=–0.42, N=12, p=0.20) or the weekly expenditure (r=0.26, df=12, p=0.43). No significant associations were noted when the same correlations were performed for VMAT2 availability in the functional or anatomical subdivisions of the striatum.
Discussion
In this human imaging study, we investigated VMAT2 availability in a group of subjects who regularly abused cocaine for nearly two decades, and we confirmed lower VMAT2 availability (10.0% to 16.3%) in the cocaine-dependent subjects than in matched healthy comparison subjects. The results of this study are in agreement with those from three of four studies that previously evaluated this issue in postmortem brain tissue and showed comparably lower VMAT2 (12% to 22%) in chronic cocaine abusers than in comparison subjects. They are also in agreement with the results of one (
24), but not another (
25), [
11C]DTBZ PET study that evaluated this issue in methamphetamine abusers. The finding in the study by Boileau et al. (
25), which demonstrated 10%–22% greater VMAT2 binding in the subdivisions of the striatum in methamphetamine abusers than in healthy comparison subjects, is inconsistent with both the results of a previous study by Johanson et al. (
24) of methamphetamine abusers and the results of this study of cocaine abusers, showing lower VMAT2. A possible reason for the paradoxically greater VMAT2 binding in methamphetamine dependence that was reported by Boileau et al. is related to the scanning of a relatively large proportion of the methamphetamine abusers shortly after the cessation of drug use (eight of the 14 methamphetamine-dependent subjects tested positive for methamphetamine and/or cocaine on the day of the PET scan), as opposed to a longer period of abstinence. Thus, it is likely that the evaluation of VMAT2 binding in their study was influenced by methamphetamine-induced transient alterations in dopamine concentration, as demonstrated by these authors in follow-up investigations (
26,
27). In our study and the study by Johanson et al. (
24), the interaction between cocaine- or methamphetamine-induced transient alterations in dopamine and VMAT2 was less of an issue, as all subjects had been abstinent from drugs for a minimum of 14 days before the PET scan. Nevertheless, as shown in
Figure 2, data from this study indicate an overlap in [
11C]DTBZ BP
ND between the cocaine abusers and comparison subjects, which may be due to various demographic and clinical factors, such as social status, the duration and amount of cocaine abuse, or genetic polymorphisms—all of which need to be investigated in a larger cohort.
It is not possible to ascertain from this study whether the lower VMAT2 in cocaine addicts reflects a compensatory down-regulation of presynaptic dopamine storage vesicles, a loss of dopaminergic terminals, or a combination of both. A down-regulation of vesicular dopamine stores may suggest a compensatory mechanism that counteracts the repeated release of dopamine into the synapse that is caused by chronic cocaine abuse. Alternatively, the loss of dopamine terminals would suggest a more permanent cocaine-induced neurotoxicity that leads to reduced dopamine release. Future studies in cocaine addicts are necessary to determine whether there is recovery of [11C]DTBZ binding potential to the levels observed in healthy subjects with a more prolonged duration of abstinence, such as 6 to 12 months. Such a recovery would indicate a compensatory mechanism, as opposed to neurotoxicity of chronic cocaine abuse in humans.
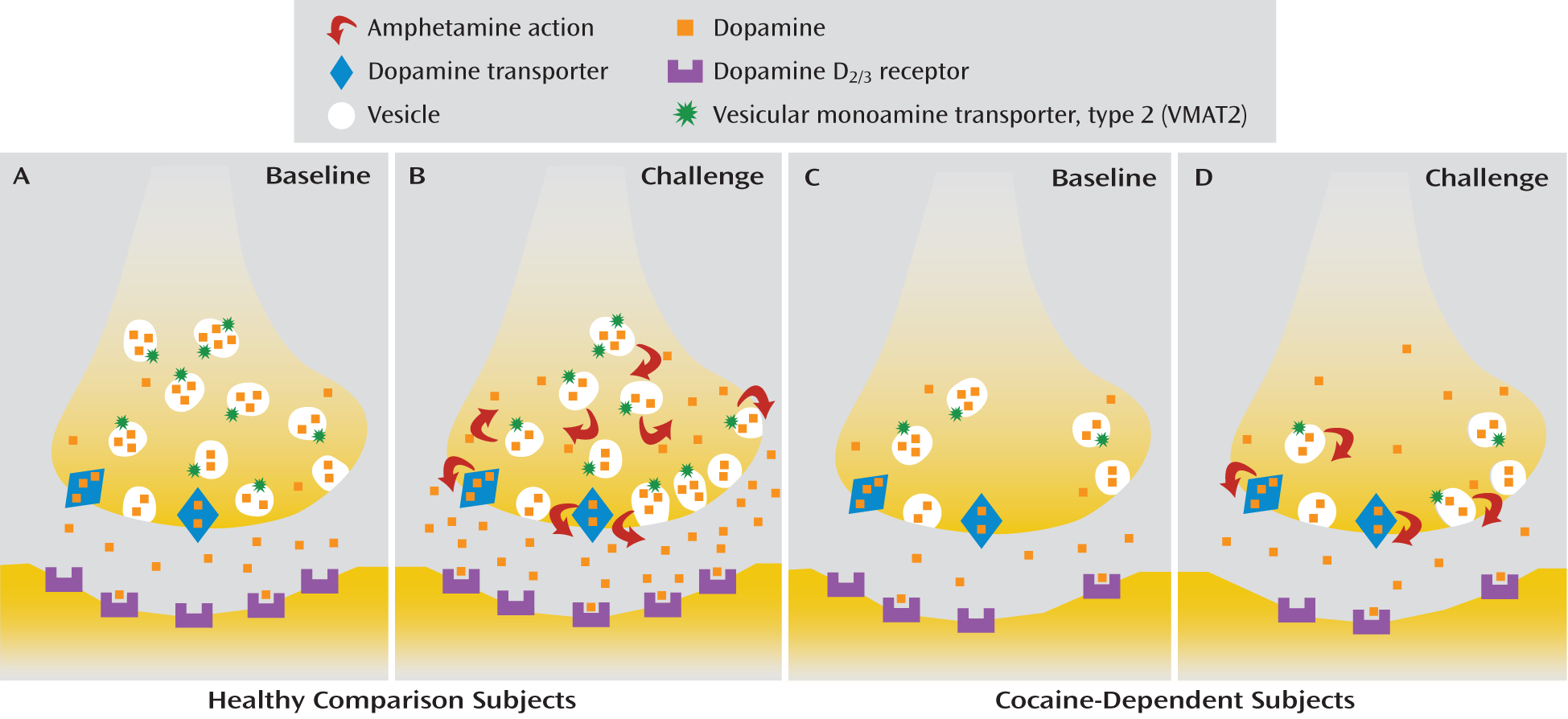
The blunting of stimulant-induced dopamine release in cocaine abusers (
Figure 3) is one of the most robust and replicated findings in the addiction imaging literature (
1,
2,
4,
28). As more recent studies in laboratory and clinical settings also suggest that this phenomenon is associated with relapse in cocaine addicts, it is critical to understand the mechanisms contributing to lower dopamine release in order to advance therapeutics for addictive disorders (
2,
4). Two possibilities have been identified and discussed in the literature with respect to the mechanism of action of amphetamines (and to a lesser extent other stimulants, such as methylphenidate): vesicular depletion, the process by which amphetamine displaces dopamine from secretory vesicles into the neuronal cytoplasm (
29), and reverse transport, the process by which cytoplasmic dopamine is released into the extracellular space through outward transport by the dopamine transporter (
30). The relative importance of these two processes, vesicular depletion and reverse transport, with respect to the amount of dopamine released following an acute amphetamine challenge has been debated extensively in the literature (
31–
33). Nevertheless, preclinical studies clearly show that the number (B
max) of dopamine storage vesicles and dopamine transporters affects the amount of dopamine released after an acute amphetamine challenge (
34,
35). On the basis of these data we hypothesized that a decrease in available dopamine storage vesicles and/or dopamine transporters following chronic cocaine use leads to blunting of amphetamine-induced dopamine release in cocaine dependence. In a review of the literature on this topic, we found little evidence to support lower than normal dopamine transporter availability in cocaine abusers (
6). Seven of the 10 postmortem studies that contrasted dopamine transporter binding in cocaine abusers and comparison subjects supported greater binding in the cocaine abusers, two studies showed no difference, and one showed lower binding (reviewed in Table 4 of reference
6). The in vivo PET studies that had evaluated this issue were also split—with one suggesting a modest up-regulation of dopamine transporter during early withdrawal (<96 hours) (
36) and the other suggesting no change after 3 to 12 weeks of abstinence (
37). In contrast to the data on the dopamine transporter, the in vitro literature and this PET study support lower VMAT2 availability in cocaine abusers. As VMAT2 both directly regulates the size of the vesicular dopamine pool and indirectly influences the amount of dopamine that is available in the cytosol for carrier-mediated dopamine release (reverse transport of dopamine transporter), it is likely that lower VMAT2 leads to reduced stimulant-induced dopamine release in cocaine addicts. Investigators in future studies should attempt to understand the clinical relevance of lower VMAT2 to relapse and outcome in abstinent cocaine abusers.
Finally, in contrast to the postmortem and imaging data from cocaine abusers, no such evidence of lower [
3H]DTBZ binding has been found in chronically cocaine-treated rodents (
38,
39). This discrepancy in VMAT2 binding between the human and rodent data on chronic cocaine use may explain the paradoxical findings observed with regard to acute amphetamine challenge across species. In human cocaine abusers, low dopamine release is observed, whereas studies of rodents repeatedly exposed to cocaine demonstrate greater dopamine release following an acute amphetamine challenge (termed “sensitization,” reviewed in reference
6). These data suggest that perhaps chronic and repeated exposure to cocaine leads to loss or death of dopaminergic nerve terminals in humans, but not in rodents, which are typically exposed to cocaine for a shorter time (weeks, compared to decades in humans) in the laboratory. Future investigations need to investigate whether relatively modest reductions in presynaptic dopamine nerve terminals (measured as 10% to 20% reductions in VMAT2 binding) lead to profound reductions (60% to 90%) in stimulant-induced dopamine release, as observed in cocaine-abusing humans relative to healthy comparison subjects (
2).
In conclusion, we found that repeated exposure to cocaine is associated with lower VMAT2 availability in the striatum of cocaine abusers. This lower striatal VMAT2 availability, which leads to lower vesicular dopamine in the presynaptic terminals, may be one of the several mechanisms that contribute to the lower dopamine release in the brain's reward circuit and thereby drive relapse in cocaine addicts. Further research is necessary to understand the clinical significance of this finding in a much larger group of cocaine-dependent subjects.