Attention deficit hyperactivity disorder (ADHD) is characterized by age-inappropriate symptoms of inattention, impulsiveness, and hyperactivity (
1). It affects 3%–8% of school-age children, disrupting academic and social development, and persists into adulthood in 65% of those affected, amounting to 4% of the adult population (
2,
3). Cognitively, children and adults with ADHD have deficits in late-developing cognitive functions, most prominently inhibition, attention, motivation, and timing functions that are known to be mediated by late-developing frontostriatal and cerebellar networks (
4,
5).
The biological origins of ADHD are complicated by heterogeneous clinical symptoms, comorbidity (in approximately 65% of the cases) with other disorders (conduct, mood, and anxiety disorders and Tourette's syndrome), and exacerbation by adverse environments and psychosocial events (
6). The last two decades of structural and functional imaging studies have shown that ADHD is associated with deficits in the structure, functioning, and connectivity of frontostriatal, parietotemporal, and frontocerebellar networks (
5,
7,
8). A recent meta-analysis of all structural voxel-based morphometry studies in ADHD, however, showed that the most consistent deficit is low gray matter volume in the basal ganglia (
9).
Studies using positron emission tomography (PET) and single photon emission tomography (SPECT), conducted mostly in adults with the disorder, have focused predominantly on the neurotransmitter dopamine, given that it is known to have a central role in the regulation of psychomotor activity, motivation, and the frontostriatal-mediated inhibitory, timing, and attention functions that are compromised in the disorder (
10). The focus on striatal dopamine has been reinforced by structural, functional, and neurochemical imaging findings of striatal deficits (
5, 7–
9). The striatum receives dense ascending projections from the mesencephalic dopamine neurons of the substantia nigra and ventral tegmental area (
11). The interest in striatal dopamine was further heightened by the fact that the striatal dopamine transporter is the main target for one of the most effective treatments of ADHD, methylphenidate (
12–
15), which has been shown to block about 70% of striatal dopamine transporters (
16), leading to enhanced striatal dopamine availability (
12). Last, dopamine receptor and transporter genotypes have been associated with the disorder (for a meta-analysis, see reference
13).
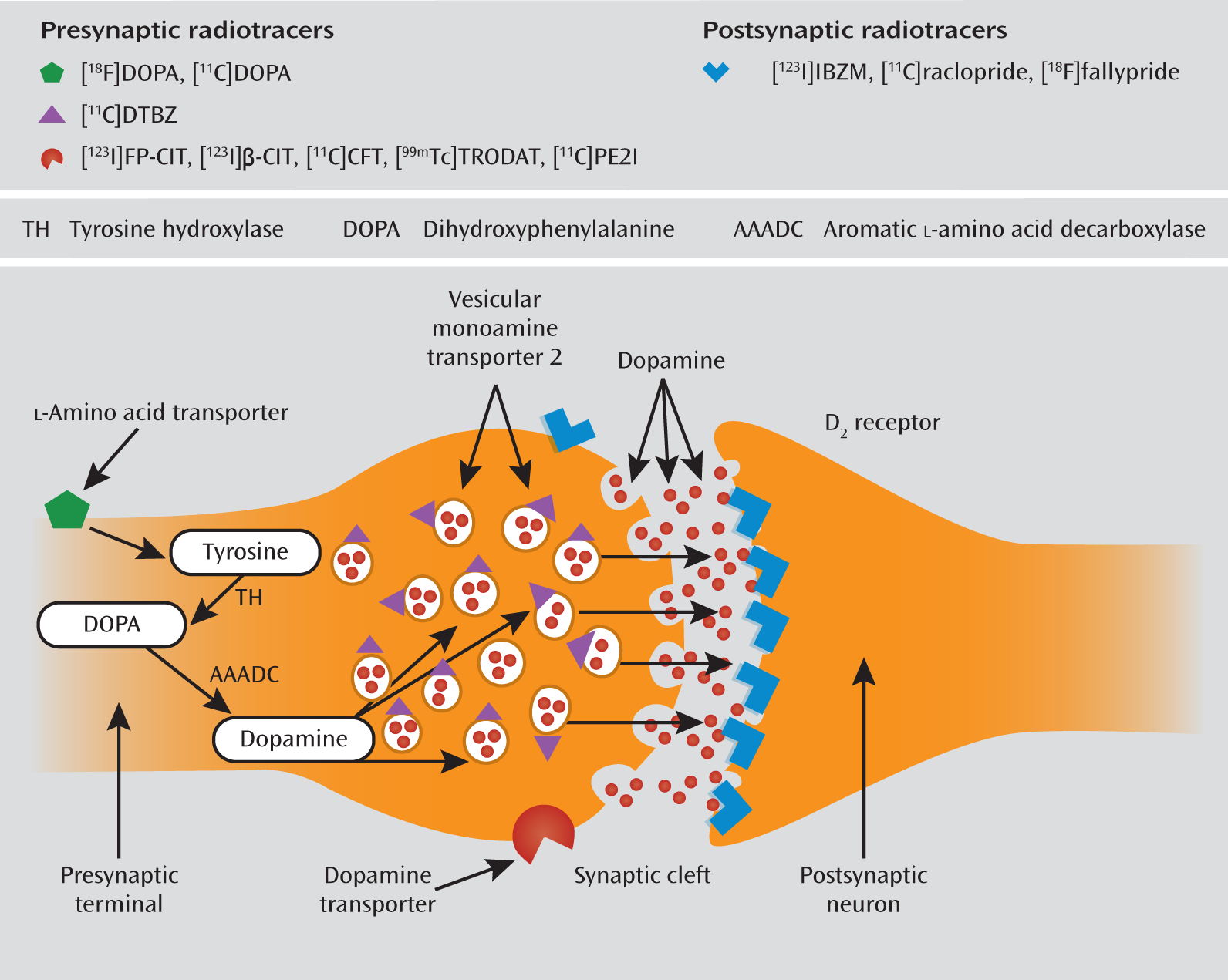
The membrane-bound presynaptic dopamine active transporter plays a key role in regulating the dopamine content in the synaptic cleft by removing the dopamine molecules from the synaptic cleft and returning them to the presynaptic cell (16a [available
here]). Dopamine transporters are localized in dopaminergic axons, with the highest levels in the striatum and olfactory tubercle and much lower levels in the amygdala, hypothalamus, hippocampus, thalamic nuclei, and neocortex (
14). Because striatal dopamine transporters are located exclusively on dopamine-synthesizing neurons, the measurement of striatal dopamine transporter density is a specific marker of presynaptic dopaminergic neuron integrity (
15). It is possible to simultaneously visualize and quantify dopamine transporter binding by using radioligands and SPECT or PET (
Figure 1).
The findings on dopamine transporter levels in the striatum of patients with ADHD relative to those of comparison subjects have been inconsistent. While several of the earlier studies showed higher dopamine transporter levels in ADHD patients (
16), some showed no difference (
17) and others indicated lower dopamine transporter levels in ADHD (
10). Reasons for the discrepancies could be 1) differences in radiotracers or the methods used, 2) differences in patients' characteristics, including medication history, comorbid conditions, and age, and 3) differences in study group sizes. One of the most compelling confounds is psychostimulant medication, given the known acute effect of psychostimulants of modulating striatal dopamine transporters (
12). A number of critical reviews have addressed dopamine transporter alterations in ADHD (
6,
18–
21); however, to our knowledge, no formal meta-analysis has ever tested the magnitude of these abnormalities while controlling for the aforementioned confounds, particularly medication effects.
In this meta-analysis of PET and SPECT studies, we aimed to examine the meta-analytic evidence for a consistent alteration of striatal dopamine transporter density across studies. In addition, we aimed to assess the effect of medication history and a number of other moderator variables, including age, comorbid conditions, gender, and publication year.
Discussion
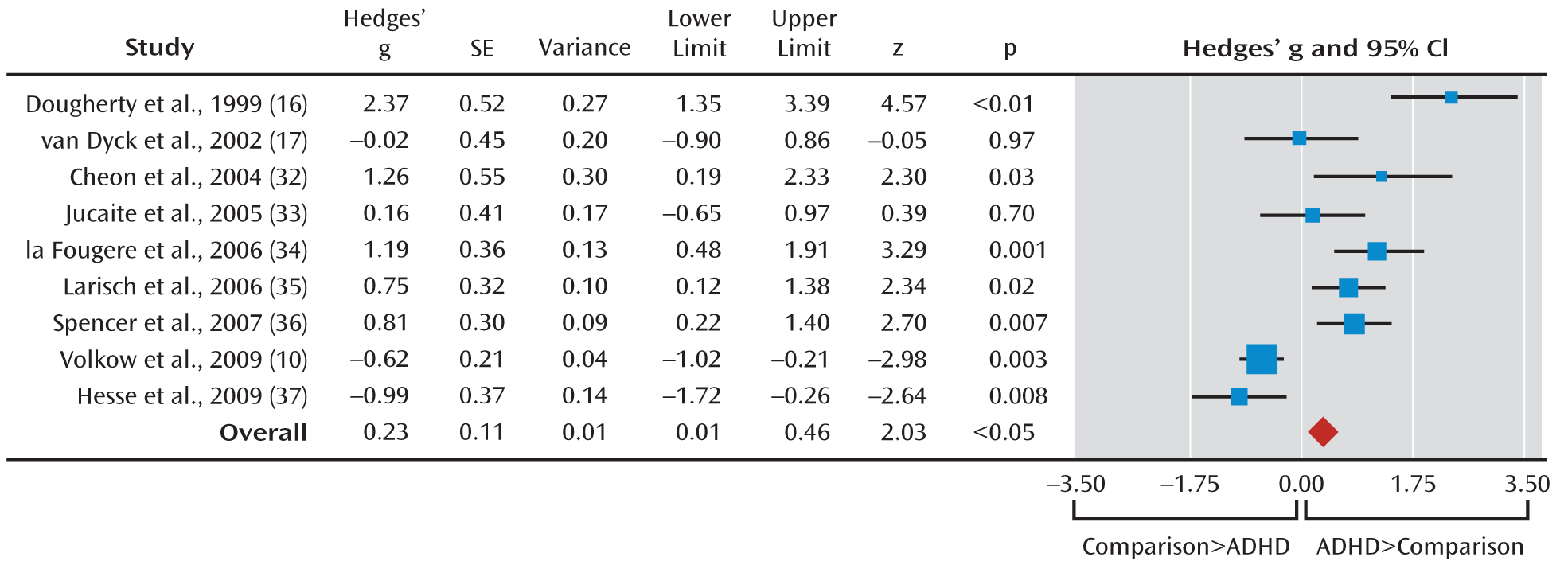
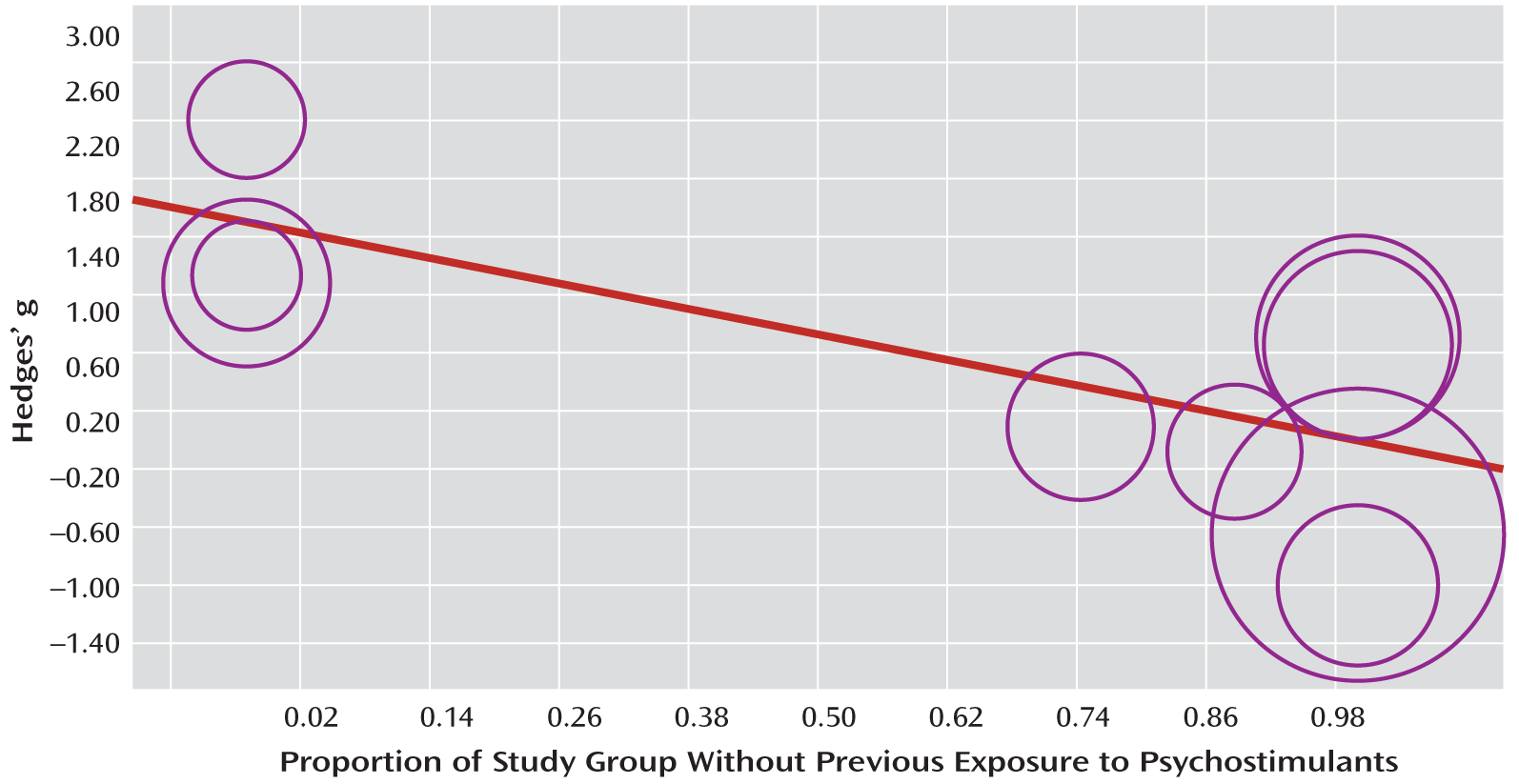
To our knowledge, this is the first comprehensive meta-analysis addressing striatal dopamine transporter density in ADHD. Meta-analytic evidence from approximately 170 ADHD patients showed on average 14% greater density of striatal dopamine terminals in the ADHD patients than in healthy comparison subjects. However, the effect size was small and heterogeneity across studies was substantial, with a prominent effect of psychostimulant history on the findings, accounting for a substantial proportion of variance across studies. Thus, the meta-regression analysis showed that the percentage of patients without a medication history was negatively associated with dopamine transporter level, so that studies of patients receiving long-term medication showed higher dopamine transporter levels, while an absence of medication exposure was associated with lower dopamine transporter levels relative to the healthy comparison subjects. Conversely, age, gender, comorbid conditions, year of publication, and type of imaging technique had no effect on the meta-analytical estimates of striatal dopamine transporter density.
The negative correlation accounted for about half of the overall variance across studies. This suggests that the higher striatal dopamine transporter density in ADHD may be the consequence of previous treatment with stimulants and hence be the result of an adaptive response of the brain to the continuous dopamine transporter blockade with psychostimulants. This suggests that a high dopamine transporter level is not part of the key ADHD pathophysiology but is secondary to years of psychostimulant treatment and reflects an adaptive brain response to the long-term blockade of dopamine transporters by psychostimulants. This notion of an adaptation to psychostimulants is in line with the finding that methylphenidate is effective in improving clinical symptoms of ADHD in the short term but that long-term effectiveness is limited; larger doses are often required to maintain clinical effectiveness, and clinical effectiveness appears to wane after years of medication (
39). A caveat is that these findings are from cross-sectional analyses, with a selection bias, and we therefore cannot infer direct causality. The theory of an adaptive response of the brain to psychostimulant medication would have to be tested directly in longitudinal imaging studies using a randomized controlled design.
The meta-analytical finding of lower dopamine transporter levels in medication-naive patients is in line with the findings from the study by Volkow et al. (
10), which was one of the largest weighted studies in our meta-analysis and one of the best-controlled studies, enrolling a large number of drug-naive ADHD subjects with no comorbid psychiatric or neurological conditions and adopting stringent inclusion criteria to control for past ADHD medication and/or drug abuse history. The finding of lower striatal dopamine transporter levels in medication-naive patients is also consistent with the prominent theory that ADHD is a dysfunction of dopamine neurotransmission, with a consequent dysregulation of dopamine-modulated circuits. In particular, the striatum appears to play a prominent role in ADHD symptoms (
12).
In normal development, a 6%–8% decline in striatal dopamine transporters has been observed per decade in PET and SPECT studies (
40). We observed nonsignificantly lower striatal dopamine transporter levels in older relative to younger adults in the comparison group but no difference in the patients. This may hint at abnormalities in the normal age-associated striatal dopamine transporter development in ADHD patients, or it may be related to stimulant treatment.
Methylphenidate hydrochloride is one of the most efficacious treatments for ADHD, reducing symptoms in up to 70% of children (
12). PET studies have shown that methylphenidate blocks dopamine transporters in the striatum in a dose-dependent fashion (
41,
42), leading to an increase in extracellular striatal dopamine (
43). The amount of extracellular dopamine (
44) released by psychostimulants, however, is likely to depend on a combination of the blockade of dopamine transporters and the baseline rate of dopamine release, which is regulated by individual differences in dopamine cell firing and by environmental stimulation (
12). Studies using [
18F]DOPA or [
11C]DOPA PET (
43,
45) confirmed low dopamine synthesis capacity in the striatum of ADHD patients. Studies using [
11C]raclopride PET to investigate postsynaptic receptor binding further showed that dopamine activity is depressed in ADHD, supporting the dopamine deficit theory of ADHD (
46). Given that dopamine signals the saliency of stimuli and drives the motivation to perform goal-directed behaviors, the methylphenidate-induced amplification of the striatal dopaminergic signal would cause increased saliency perception, motivating the individual to engage and improving attention and performance (
12).
Our meta-analysis finding that previous treatment with psychostimulants increased striatal dopamine transporter density in ADHD patients may seem counterintuitive, given the dopamine transporter blockade by methylphenidate. Enhanced striatal dopamine transporter density in patients receiving long-term treatment, however, could reflect a secondary adaptive brain response to the chronic striatal dopamine transporter blockade, i.e., adjustment to chronically elevated striatal dopamine availability through up-regulation of dopamine transporter levels. There is evidence in favor of this theory from a small prospective study, which showed that after 1 year of stimulant medication the striatal dopamine transporter levels of ADHD patients in fact increased (
47). Furthermore, there is evidence indicating that cocaine—which is a stimulant drug that, like methylphenidate, blocks dopamine transporters—not only blocks the acute regulatory effect of the dopamine transporter on synaptic dopamine levels but also may exert the opposite effect: insertion of dopamine transporters from the endosomic recycling pool into the plasma membrane (
48). In rhesus monkeys, chronic administration of cocaine up-regulates striatal dopamine transporter expression, which persists for more than 30 days after cocaine withdrawal (
48). High dopamine transporter expression has also been shown in postmortem analyses of brain tissue from human cocaine addicts, and synaptosomes prepared from this tissue exhibit greater dopamine uptake than synaptosomes from age-matched cocaine-naive individuals (
48).
Methylphenidate-associated changes have also been observed in brain function and structure. Functional MRI studies in patients with ADHD have shown that single and long-term doses of methylphenidate up-regulate and normalize typically low frontostriatal brain activation (
49–
54). Because striatal dopamine transporters are located exclusively on dopamine-synthesizing neurons, their measurement is a specific marker of dopaminergic neuron integrity in the basal ganglia. The notion of adaptive brain changes in response to long-term psychostimulant treatment is also in line with the results of several structural imaging studies showing that patients with ADHD receiving long-term medication have more normal structure and morphometry in the basal ganglia than medication-naive patients (
55,
56). These findings are further confirmed by a recent meta-analysis of whole-brain structural imaging studies, which showed that the lower basal ganglia gray matter volume in ADHD patients, relative to that in comparison subjects, is dependent on long-term medication (
9). The percentage of patients receiving long-term medication was linearly associated with basal ganglia size, such that studies in which more than 70% of the patients were medicated did not show striatal abnormalities, while the greatest deficits were observed in studies of medication-naive patients.
It is, however, also possible that lower dopamine transporter density and lower dopamine release in medication-naive ADHD patients reflect prefrontal pathology, well demonstrated in neuroimaging results for ADHD (
5), since frontostriatal glutamatergic circuits regulate striatal dopamine release.
The present study has several limitations. The meta-regression finding of an association between medication and dopamine transporter density is limited by the cross-sectional nature of the analysis, and causality of the regression findings needs to be established in longitudinal prospective studies using a randomized controlled design. It has been suggested that high heterogeneity across studies may be due to differing dopamine transporter sensitivity across the different radiotracers employed. For example, there is evidence that the specific-to-nonspecific ratio of labeled cocaine is relatively low and that this radioligand may occupy binding sites other than those occupied by FP-CIT, altropane, TRODAT-1, and IPT (
57). Furthermore, dopamine transporter binding may be influenced by a complex network of interactions with other receptors or neurotransmitters. For example, there is recent evidence that norepinephrine transporters contribute to the pathophysiology of ADHD, with norepinephrine transporter blockade in frontal regions underlying some of the therapeutic effects of methylphenidate (
58). However, while methylphenidate enhances both norepinephrine and dopamine in prefrontal brain regions, where it blocks both the relatively densely distributed norepinephrine transporters and the less densely distributed dopamine transporters (
58), in the basal ganglia methylphenidate has minimal effects on norepinephrine levels, since the norepinephrine transporter density is vanishingly low (
6). Other studies have found that presynaptic D
2 autoreceptor activation, normally constraining dopamine action at synapses, regulates dopamine transporter activity that modulates synaptic dopamine homeostasis (
59). The picture is further complicated by a potential interplay between dopamine transporter functioning and nicotinic neurotransmission at the presynaptic level (
60). Finally, there is recent evidence suggesting that the ADHD diagnostic category comprises multiple entities with different underlying pathophysiologies and abnormalities of neurotransmitter profiles (
61). In particular, children with the combined/hyperactive subtype of ADHD show a higher frequency of conduct disorder and good response to treatment, are exposed to more moderate stress during their mothers' pregnancies, and have a higher frequency of the L genotype for a polymorphic region of the serotonin transporter gene, as compared to children with the inattentive ADHD subtype (
61). On the basis of animal studies showing dopamine transporter differences in the pathophysiology of the two subtypes, it is possible to speculate that treatment with methylphenidate might normalize abnormal dopamine transporter levels more effectively in the combined subtype (
62).
The meta-analysis and meta-regression analysis show that striatal dopamine transporter levels in ADHD depend on chronic psychostimulant treatment, so that medication-naive patients have low striatal dopamine transporter levels, whereas patients receiving long-term medication have high levels. Consequently, the previously reported high dopamine transporter density in ADHD patients may potentially represent up-regulation secondary to chronic administration of psychostimulants, rather than primary pathophysiology of ADHD. Future prospective studies using randomized controlled trials in large groups of drug-naive individuals with ADHD will be needed to definitively clarify the long-term effect of psychostimulants on striatal dopaminergic neurotransmission.