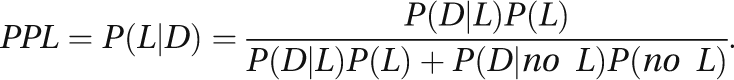The CAPS project is specifically funded to reanalyze HGI data using the posterior probability of linkage (PPL) framework as implemented in the software package KELVIN (
1). There are three principal advantages to using the PPL for linkage analysis of the HGI data (see the Method section for details). First, it is essentially model free, while retaining the advantages of likelihood-based analyses, making full use of all available data, including from unaffected individuals. Second, it is specifically tailored to handle multiple, potentially highly heterogeneous data sets or subsets, using Bayesian sequential updating to accumulate linkage evidence across subsets while allowing explicitly for genetic differences between subsets. And third, in stark contrast to p values or maximum LOD scores, based on either “mega-analysis” or traditional meta-analysis, the PPL can accumulate evidence both for and against linkage at each genomic position. This increases resolution of a genome scan by excluding stretches of the genome in an essentially model-free manner, while also distinguishing common as opposed to distinct genetic features across different populations or clinical subgroups.
The PPL is thus uniquely well suited to the analysis of multisite, potentially highly heterogeneous, genetic data. Here we consider omnibus linkage results, based on all of the HGI schizophrenia family data in aggregate. We also consider population-specific findings and the possibility of specific loci underlying a clinical schizophrenia subtype involving affective components.
Results
We first present omnibus linkage results, considering all of the data in aggregate. We then consider common and distinct loci, by major population group and by clinical subgroup (families with narrow schizophrenia only versus families containing at least one case of a schizophrenia/affective diagnosis). We then compare our results with results from the original publications for studies 1–7, as well as with results based on independent sets of data and meta-analyses.
Omnibus Results
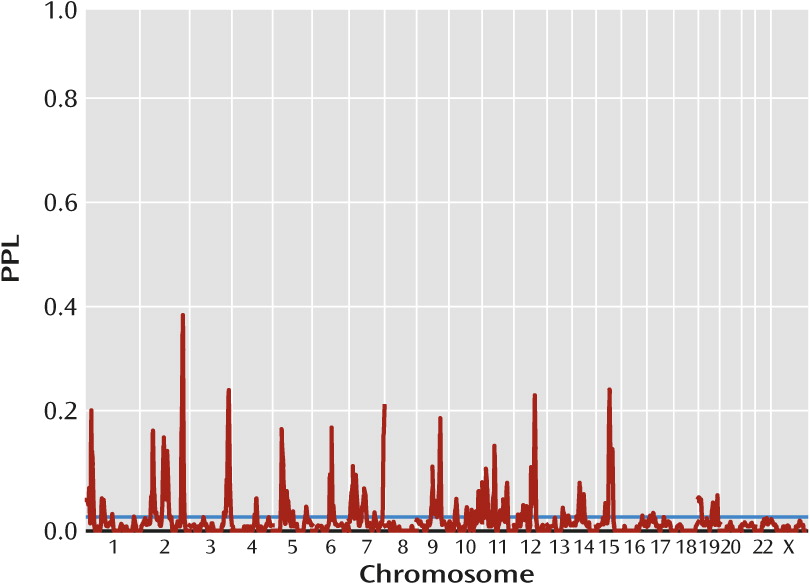
Figure 1 presents omnibus linkage results, sequentially updated across all data subsets. Overall, 76% of the genome showed evidence against linkage (PPL<0.02), while only 10% showed PPL>0.05 and 4% showed PPL>0.1. There were twenty distinct loci with PPL>0.1, 13 with PPL>0.15, and four with PPL>0.25. These last occurred at 2q36.1 (PPL=0.41), 3q28 (PPL=0.27), 12q23.1 (PPL=0.26), and 15q23 (PPL=0.27) (
Table 1).
Population-Specific Results
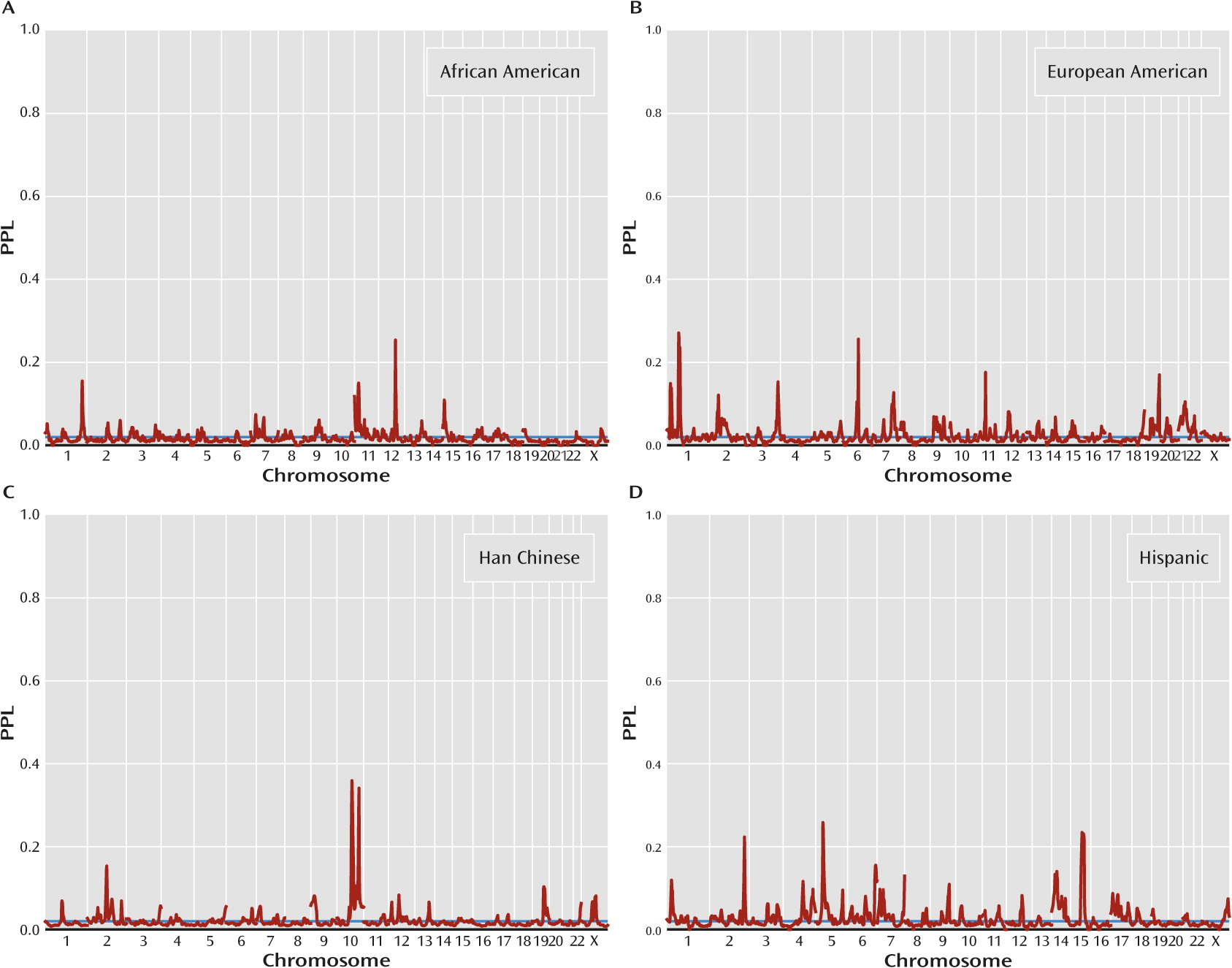
Figure 2 presents population-specific results. There are four population-specific loci with PPL>0.25 (
Table 1). Two of these come from Han Chinese (10q22.3, PPL=0.36; 10q26.12, PPL=0.34), and at both these loci all three of the other population groups show evidence against linkage. Similarly, European American data support 1p32.3 (PPL=0.27), with data from all other population groups providing evidence against linkage at this locus. By contrast, the Hispanic-specific peak at 5p14.1 (PPL=0.26) receives modest support from African American data (PPL=0.04) but evidence against linkage in the other two sets. The four omnibus peaks (see above) are each supported by at least three population groups, in each case with the remaining group being neutral, that is, showing evidence neither for nor against linkage (
Table 1).
Clinical Subset-Specific Results
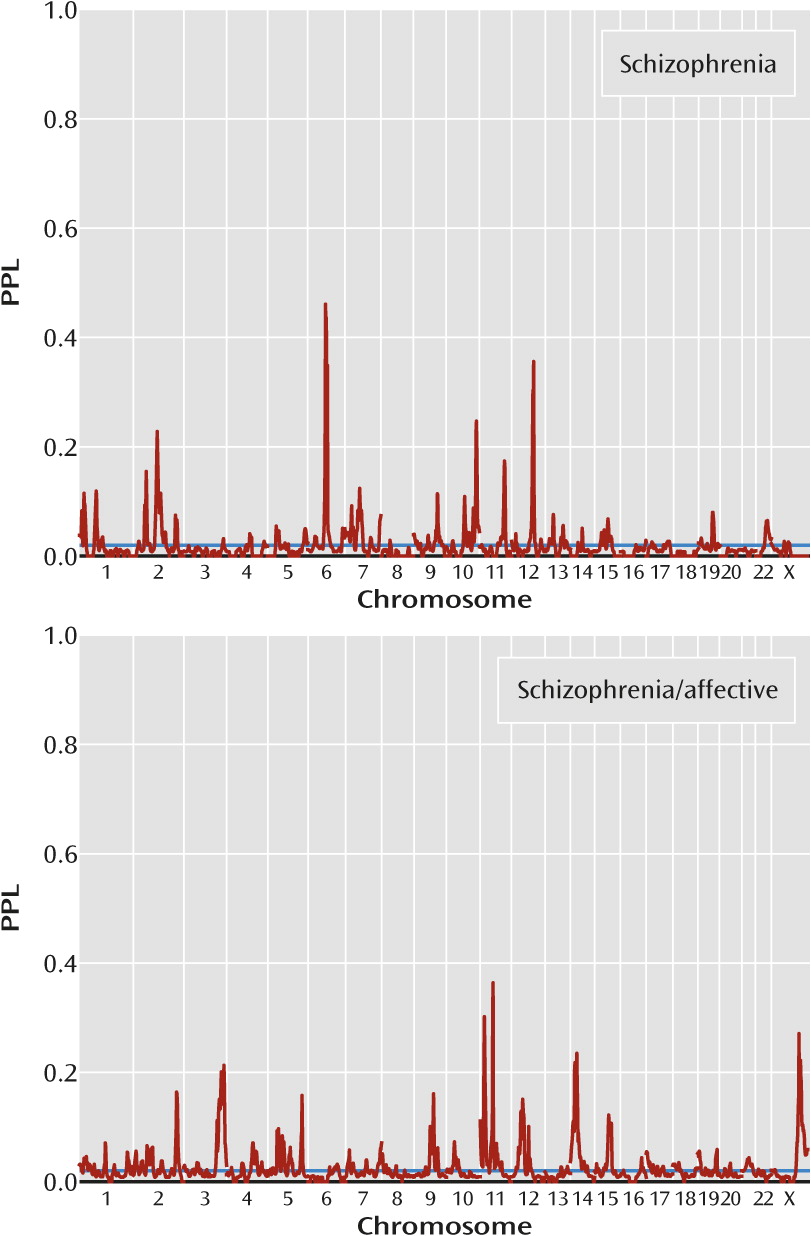
Figure 3 presents results by clinical subgroup. There are five loci with PPL>0.25 in either of the two clinical subgroups (
Table 1), and in every case the other subgroup provides evidence against linkage (in each case, PPL<0.01). Notably, three of these peaks (11p15.3, 11p11.2, 23q26.1) are supported by the far smaller schizophrenia/affective subset, suggesting that this really does constitute a more homogeneous classification of the families. However, there is possible confounding by population group here, since each clinical subset includes all four population groups, but in differing proportions (see the Discussion section).
Comparison With Original Study Results
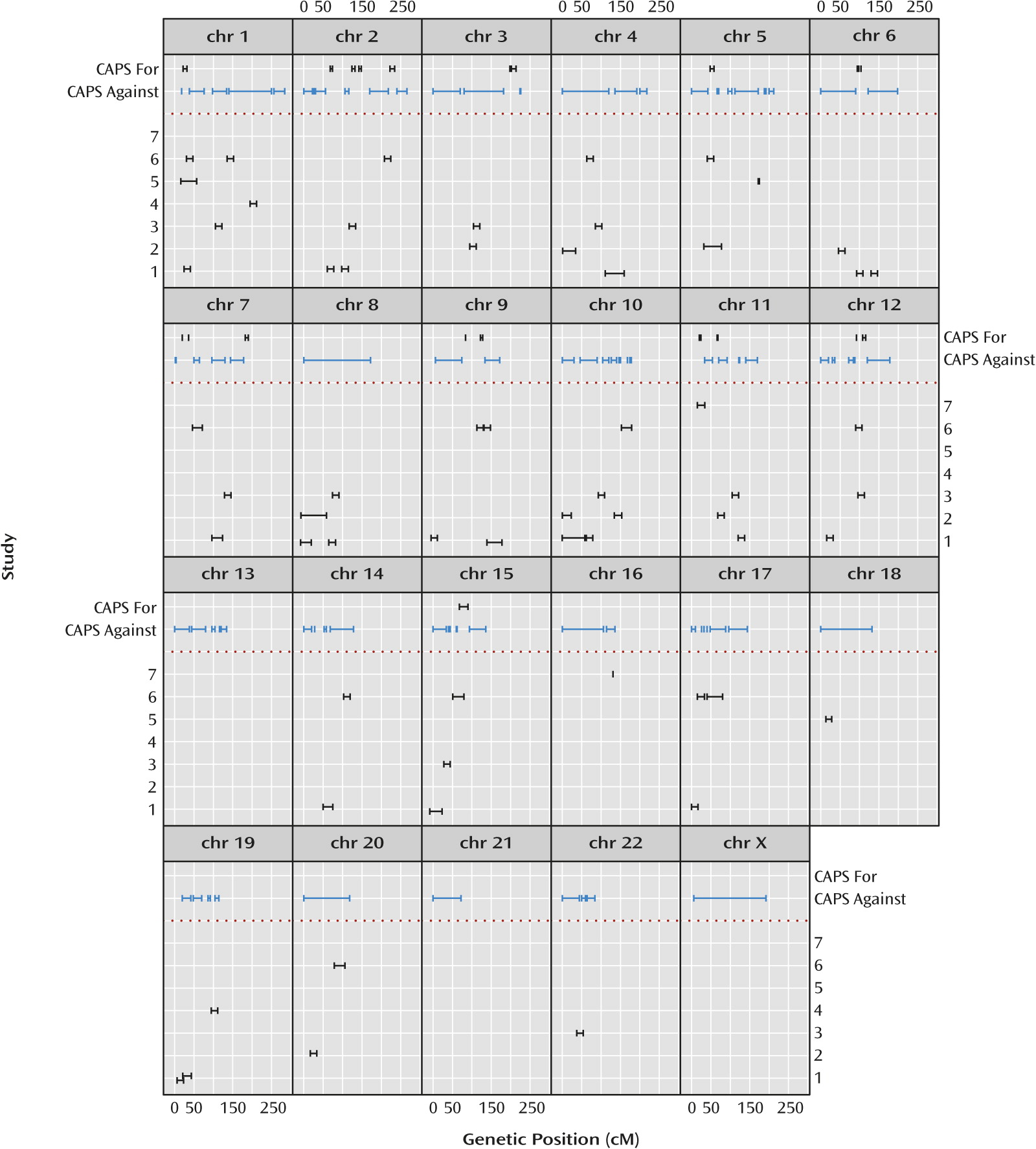
Figure 4 illustrates salient differences between the omnibus results and results extracted from the original published linkage analyses of the individual studies. The 10% PPL threshold was chosen based on qualitative visual separation of salient signals from background noise in
Figure 1 and is intended to mimic the criterion of “suggestive” evidence as shown in the figure based on the original publications. There is no rigorous way to precisely compare the magnitude of results across data-analytic methods or to derive and apply exactly comparable interval estimates. Thus, the figure is intended to convey in broad strokes differences in the overall genomic landscape between the individual original reports and our omnibus analyses rather than to present precise quantitative differences.
Of the 20 intervals with PPL>0.10, 12 line up in or near an originally reported interval. This could be coincidental, however, since all but three chromosomes are implicated (generally at the level of “suggestive” linkage) by at least one of the original publications, with little overlap in reported loci across the original studies.
Figure 4 also shows the large portions of the genome showing evidence
against linkage using the PPL. The PPL’s ability to accumulate evidence against linkage has no correlate in either the original nonparametric linkage or meta-analyses.
Comparison With Results From Independent Studies
Of the four omnibus loci with PPL over 25%, all are in regions of previous schizophrenia linkage reports. Chromosome 2q36.1 is in a region that has been implicated in several previous linkage reports, including a significant finding in a Finnish sample (
23), and has received suggestive support from other reports (
5,
24,
25) that include families found in study 2 and study 3.
SCG2, the gene encoding secretogranin II, is located under the center of this peak and has been reported to have increased expression in the dorsolateral prefrontal cortex in individuals with schizophrenia (
26). Chromosome 3q28 is supported by suggestive linkage results in studies in Finnish (
23), Palauan (
27), and Dagestani (
28) samples that are not part of the NIMH-HGI collection. Chromosome 12q23.1 has been implicated by a suggestive linkage report with a negative symptom factor score (
29). While the 12q candidate gene
DAO is located beyond the boundaries of this peak, in an area where the omnibus analysis produced evidence against linkage, the gene
PAH is located near the edge of the omnibus peak, in a region with PPL scores slightly above 2%.
PAH, the gene for phenylalanine hydroxylase, while traditionally associated with phenylketonuria, has been suggested as a schizophrenia candidate gene in multiple studies (
29–
33). Of further interest is the 12q linkage peak observed in the African American subset; this peak has a maximum PPL score of 25.4% located 4 centimorgans (cM) telomeric to the omnibus peak, very near
PAH. The omnibus peak on chromosome 15q23 overlays a suggestive linkage peak that was originally reported in the Hispanic samples included in this analysis (
8).
Four additional peaks over 25% were observed in specific population groups. The 10q22.3 and 10q26.12 peaks in the Han Chinese sample overlap the linked regions from the original report on these families (
5). Similarly, the Hispanic-specific peak at 5p14.1 overlaps a suggestive linkage peak that was previously reported in these families (
8). By contrast, the 1p32.3 locus seen in the European American subset was not suggested by the individual studies contributing to this subset (
4,
34,
35). Having data from one population group provide evidence for linkage and data from the other groups provide combined evidence against is consistent with either separate sets of genes operating in the different population groups or, more likely, dependence of the salience of genetic effects on the background (ancestral) genome or environment. It is also consistent, however, with the subset-specific PPLs being overestimates of the actual probability of linkage, which is given by the omnibus PPL. Differences in sample sizes across groups (139 African American, 187 European American, 483 Han Chinese, and 161 Hispanic) also complicate interpretation.
Dividing the data by clinical subgroup (families containing individuals with schizophrenia/affective diagnoses versus families with only narrow schizophrenia) yielded four additional loci with PPL>25%, and in every case the locus was supported by data from one group while data from the other group provided evidence against linkage. This strongly suggests that the genetics of schizophrenia in families characterized by the presence of schizoaffective disorder or schizophrenia with a comorbid affective disorder may be distinct from the genetics of schizophrenia in individuals without a significant mood disorder. On the other hand, there is considerable confounding by population, since the preponderance of families with individuals with a schizophrenia/affective diagnosis varies by group as well. The proportions of groups (African American, European American, Han Chinese, Hispanic) within schizophrenia-only families and families that include schizophrenia/affective diagnoses are 0.11, 0.15, 0.59, 0.15 and 0.28, 0.41, 0.04, 0.26, respectively.
Interestingly, however, all three locations identified in the families with schizophrenia/affective diagnoses (11p15.3, 11p11.2, and Xq26.1) have been previously implicated in linkage or association studies using samples with bipolar disorder or mixed samples with schizophrenia and bipolar disorder (
36–
40). The 11p15.3 region was also reported as a main finding in the original publication for study 7 (
9), and indeed, the African American-specific PPL reaches 15% at 28 cM. Of particular note is that study 7 reported (
9) a marked strengthening of the linkage signal at 11p15.3 when using a phenotype that included schizophrenia and schizoaffective disorder rather than schizophrenia only. Finally, the schizophrenia subset produced a peak of 46% at 6q14.1 that overlaps a peak of 26% in the European American subgroup but was not reported by any of the original European American studies. In aggregate, these results have significant implications for the inclusion of individuals with schizoaffective disorder in linkage or association studies of schizophrenia. (See also online
data supplement SA3 for a comparison with results in the National Human Genome Research Institute genome-wide association study database.)
Insofar as population is a major source of heterogeneity in these analyses, it is noteworthy that, with the exception of the Han Chinese sample, the remaining sample sizes for the different groups remain quite small. It is not surprising, therefore, that even the largest PPLs remain moderate in size. Similarly, dividing the data based on the presence of affective illness results in a far smaller subset for the group containing schizophrenia/affective diagnoses. Strikingly, however, this group also produces some of the highest PPLs. Our results suggest that both population and clinical subgroup are key classifiers for the purposes of gene mapping. Ideally, we would like to use subsets simultaneously on both criteria. However, the extremely small subsets that would result preclude this approach.
It is also of interest to compare our results with the meta-analyses of Nato et al. (
41) and Ng et al. (
42), both of which included studies from the HGI collection as well as other studies. Nato et al. identified 22 regions of interest, while Ng et al. reported eight regions with aggregate genome-wide evidence for linkage, all of which overlap with regions identified by Nato et al. While the majority of the Nato et al. (13 of 22) and Ng et al. (five of eight) regions produced some level of evidence in favor of linkage under the omnibus analysis, only two of our four omnibus peaks >25% (2q36.1 and 3q28) overlapped a region of interest from either study, with our largest peak of 41% overlapping regions of interest from both studies. Of our eight peaks >25% that were specific to an ethnic or clinical subset, only two (5p14.1 and 10q26.12) overlapped a region of interest from either study, again with the larger peak of 34% overlapping regions of interest from both studies. (See online
data supplement ST4 for detailed comparisons.)
Discussion
Our reanalyses of HGI data present a picture of the schizophrenia genome with some overlap with previous reports but also some novel loci, with differences both in terms of the specific loci implicated and in the rank ordering of these loci by relative strength of evidence. While a number of loci previously implicated using the samples from the HGI were also identified in the present analyses, such as 2q36.1 and 15q23, others were not. At the same time, several loci not previously reported in the HGI samples but supported by other reports in the literature have now been identified in the HGI collection, such as 3q28 and 12q23.1. Loci without any previous linkage reports in schizophrenia but with positional support from association studies were also identified, such as 11p11.2 and Xq26.1. Differences in the genetics of schizophrenia across population groups and in families with differing amounts of affective illness were also highlighted, in particular with 11p15.3, 11p11.2, and Xq26.1, which are prominent in the schizophrenia/affective group, overlapping with loci reported from independent studies of bipolar disorder or mixed bipolar-schizophrenia samples.
One caveat is that we employed a highly conservative approach both in deciding whom to categorize as “affected” (e.g., narrow schizophrenia with multiple exclusion criteria) and in our inclusion criteria for families (at least one case of schizophrenia plus one additional schizophrenia or schizophrenia/affective diagnosis), which led us to drop a large number of families from these analyses. While we believe that this approach is justified, particularly when attempting to make “apples to apples” comparisons across multiple different studies, we are aware that other investigators might make quite different judgments regarding these decisions. All data on the full set of families, including those we did not analyze here, are available on the HGI web site (
www.nimhgenetics.org), as are all protocols used in preparing the data files (
www.nimhgenetics.org/projects/CAPS). We hope that other investigators will find this a useful resource for carrying out their own analyses.
A second potential caveat is that we relied on linkage analysis. Given the relative paucity of validated linkage findings in psychiatric genetics, it is not unreasonable to be skeptical regarding this approach. There is a certain irony here, because past failures may be attributable in part to the fact that psychiatry was an early and enthusiastic endorser of the technique. Some of the schizophrenia data considered here date back to the 1990s, and the original studies taken individually were almost certainly underpowered, for a variety of reasons. By revisiting the HGI data and applying modern data processing and newer data-analytic methods to the existing multisite data as a whole, we believe that we have succeeded in extracting new information from older data sets of families. Of course, we have also lost something in working with the repository data, insofar as the original investigators had access to additional clinical information.
Historically there have been few gene discoveries under psychiatric linkage peaks. In this regard, too, having been a leader in methodological development put the field of psychiatric genetics ahead of its time: earlier molecular techniques were cumbersome and expensive, and combined with the width of linkage peaks in the older studies, this made gene identification under the peaks impractical. But the increasing practicality of high-throughput DNA sequencing should support gene discovery at linked loci in a manner unavailable until very recently. Indeed, human genetics is returning to a focus on “co-segregation” (i.e., linkage analysis) as a critical adjuvant to whole genome sequencing, because narrowing the search down to only those portions of the genome showing evidence of linkage dramatically reduces the amount of sequence needing to be interrogated. One could even argue that linkage analysis as a technique for gene discovery has yet to be properly applied in much of psychiatric genetics.
Even so, it might be argued that genome-wide association studies and case-control-based whole genome sequencing are a better investment than multiplex family studies. But linkage analysis and association analysis (whether to common single-nucleotide polymorphisms or rare variants) have the power to detect very different types of genetic effects: genome-wide association studies are well adapted to finding common variants conferring low relative risks in a manner that is consistent across populations (
43) (requiring independent replication ensures that effects specific to one data set are discounted [
44]). While linkage analysis is adapted to finding loci containing genes of large effect (in general, with rarer allele frequencies and much higher genotypic relative risks), and particularly when conducted using statistical methods specifically tuned for such conditions, it can find such genes even when they are causally relevant only in a subset of families (
10). One reason to continue to search for such genes is precisely because a gene discovered by linkage analysis almost certainly is a gene of large effect, albeit perhaps only in some families. Such genes may yield different insights into gene pathways and networks than genes discovered through alternative study designs. Linkage analysis is also robust to allelic heterogeneity, which can eliminate allelic associations altogether (
45).
The genetic architecture for complex disorders likely involves many different types of effects simultaneously: major gene effects in subsets of families; common background genes conferring small risks; copy number variants, perhaps particularly important in causing sporadic forms of disease; extensive locus heterogeneity, which might or might not be alleviated by refined clinical subtyping; and more complex features, including gene-by-gene and gene-by-environment interactions, epigenetics, and probably other things we have not even thought of yet. Under such circumstances, no one study design can solve all problems.
The psychiatric genetics community has an enormous resource on hand: extensive collections of multiplex families with considerable clinical information. These collections were labor intensive and expensive to amass; by contrast, the cost of genotyping or even sequencing is becoming relatively small. Returning to these collections with the benefits of hindsight seems a promising and cost-effective way to contribute to the growing understanding of genetic architecture emerging from a multitude of studies and study designs all being considered simultaneously. We have tried here to illustrate the potential of such use of retrospective data to alter and augment our understanding of the genetic underpinnings of psychiatric disorders. Of course, interpretation of the results is still hampered by the limited information content afforded by the available genotype data.
Finally, we note an important methodological issue highlighted by these results, namely, that conventional interpretations of statistical significance in terms of independent replication can lead us to overlook important loci that may have salient effects only in subsets of the data. As is well known, failure to replicate true loci is to be expected for even moderately complex disorders (
46). What is perhaps less widely appreciated is that traditional meta-analysis will tend to fail in precisely those same circumstances where independent replication cannot be relied upon for confirmation of results (
17,
18). At the same time, we are appropriately skeptical of weak findings that fail to replicate. Indeed, as reports of weak signals obtained by individually underpowered studies accumulate in the literature, and particularly in view of the failure of standard statistical methods to appropriately indicate evidence
against linkage, the overall picture of the schizophrenia genome becomes murkier, not clearer, as time goes on.
By design, repositories such as the HGI offer a solution by permitting analysis of far larger quantities of data than can be collected by any one study. However, under exactly these same conditions of underlying genetic complexity, the so-called mega-analyses, which simply pool all data into a single file for analysis, can also end up washing out important subset-specific loci by failing to appropriately allow for heterogeneity between data sets. For these reasons, we view the PPL—which does not require agreement across all data subsets, but rather accumulates the aggregate evidence both for and against linkage in a mathematically rigorous manner while allowing for differences between data sets—as particularly well suited to the task of accurately and efficiently extracting genetic information from repository collections. With the advent of affordable sequence data, we predict that revisiting family data already amassed in the HGI will provide a cost-effective mechanism not just for discovering linkage peaks, but for fine-mapping these peaks down to the level of the individual gene or variant.