Attention deficit hyperactivity disorder (ADHD) is a highly heritable early-onset neurodevelopmental disorder, with marked clinical heterogeneity, affecting males more often than females (
1–
3). It has a complex genetic architecture, as is the case for most common psychiatric disorders. Although associated rare variants have been identified (
4–
7), the general view is that the most satisfactory explanatory model of inheritance is a multifactorial, polygenic liability threshold one in which the combined effects of multiple common genetic variants with environmental factors contribute to ADHD risk.
With respect to common risk variants, there are no genome-wide significant findings for ADHD (
8,
9). Although it has been proposed that this simply reflects inadequate sample sizes (
5,
10), others suggest that the lack of findings is a consequence of psychiatric disorders (including ADHD) being explained mainly or solely by rare high-penetrance variants (
11). So far, for ADHD, rare variants in the form of copy number variants (CNVs) have been found to be associated (
4–
7), and studies have shown that other classes of rare mutations make some contribution to autism spectrum disorder (ASD) (
12–
14), another childhood-onset disorder that strongly overlaps with ADHD (
15). While the issue of the relative contributions of common and rare variants is far from being empirically resolved, pathway-analytic approaches have shown that not only do both contribute to ADHD, but they tend to act on similar functional classes of genes (
5,
16).
Given a contribution from both common and rare alleles (and presumably also alleles with intermediate frequencies), the multifactorial, polygenic liability threshold model predicts that forms of the disorder in groups of people less often affected (e.g., ADHD in females) or with more severe forms of the disorder should carry a greater genetic load, including greater enrichment of ADHD common risk alleles.
The presence of conduct disorder in youths with ADHD is known to index greater clinical severity (
17). Twin and family studies have also shown that in children with ADHD, the presence of conduct disorder symptoms indexes higher ADHD familial and genetic loading (
18–
22). For example, the relative risk for ADHD in biological relatives of probands who have ADHD with comorbid conduct disorder (relative risk=9.5) is almost double that of relatives of probands with ADHD alone (relative risk=5.4) (
18). These studies suggest that in ADHD, the presence of conduct disorder likely indexes greater genetic load. Previously, it has only been possible to infer this indirectly by measuring recurrence rates in various classes of relatives, but recent developments now allow the component attributable to relatively common alleles to be estimated using genome-wide molecular genetic data in the form of polygenic load (
23–
25). In the present study, we used ADHD polygenic risk scores derived from the largest published genome-wide association meta-analysis (
8) to test in an independent sample whether ADHD accompanied by conduct disorder is characterized by greater enrichment of ADHD “risk alleles.” We also investigated the relationship between polygenic score and conduct disorder symptoms.
Results
A total of 452 children from the Cardiff sample met inclusion criteria and had genetic and phenotypic data available. They ranged in age from 6 to 17 years (mean=10.7 years, SD=2.8); 389 were male (86.1%) and 63 were female (13.9%). The mean full-scale IQ was 87 (SD=11.2). The gender ratio and IQ scores are typical of U.K. ADHD clinic case subjects. The mean number of ADHD symptoms was 14.68 (SD=2.87; 25th percentile ADHD score=13.00, 50th percentile=15.00, 75th percentile=17.00). Within this sample, 77 individuals (17.0%) had a diagnosis of ADHD with conduct disorder and 375 (83.0%) had no comorbid conduct disorder. The mean numbers of DSM-IV conduct disorder symptoms were 3.6 (SD=1.86) and 0.5 (SD=0.76) for the ADHD groups with and without conduct disorder, respectively. There was no association between conduct disorder scores, age, and gender. In addition, 229 (50.7%) subjects met criteria for a DSM-IV diagnosis of oppositional defiant disorder; 65 of those with a conduct disorder diagnosis (84.4%) also had a diagnosis of oppositional defiant disorder. Twenty-two individuals (4.9%) had a comorbid diagnosis of anxiety disorder, and three individuals (0.7%) had a comorbid depressive disorder.
Polygenic Scores Predicting ADHD in the Target Sample
ADHD risk as defined from weakly associated alleles (N=46,156) in the discovery GWAS was significantly higher in ADHD case subjects than in comparison subjects (p=0.01) (
Table 1). Thus, as we postulated, risk for ADHD is in part attributable to common alleles tagged by the genome-wide genotyping arrays.
Polygenic Score Enrichment in Those With ADHD Accompanied by Conduct Disorder
The polygenic score representing ADHD risk was significantly higher in ADHD case subjects who had a conduct disorder diagnosis than in those who did not (p=0.01). The magnitude of the effect (as defined by R
2) was 1.1%, larger than that observed when comparing ADHD case subjects and comparison subjects (
Table 1).
To test whether our findings could be attributable to higher ADHD total symptom counts in those with conduct disorder, we tested the association between ADHD symptom count and polygenic risk score. Within ADHD case subjects, the total number of ADHD symptoms was not significantly associated with polygenic score. As expected, total ADHD score was significantly associated with total conduct disorder score (β=0.159, t=−2.900, p=0.004).
ADHD polygenic risk scores were significantly higher in female than in male case subjects (β=−0.104, t=−2.159, p=0.031), and hence all the data were reanalyzed allowing for sex as a covariate. The results were unchanged (data not shown).
Polygenic Score Predicting Conduct Disorder Symptom Scores
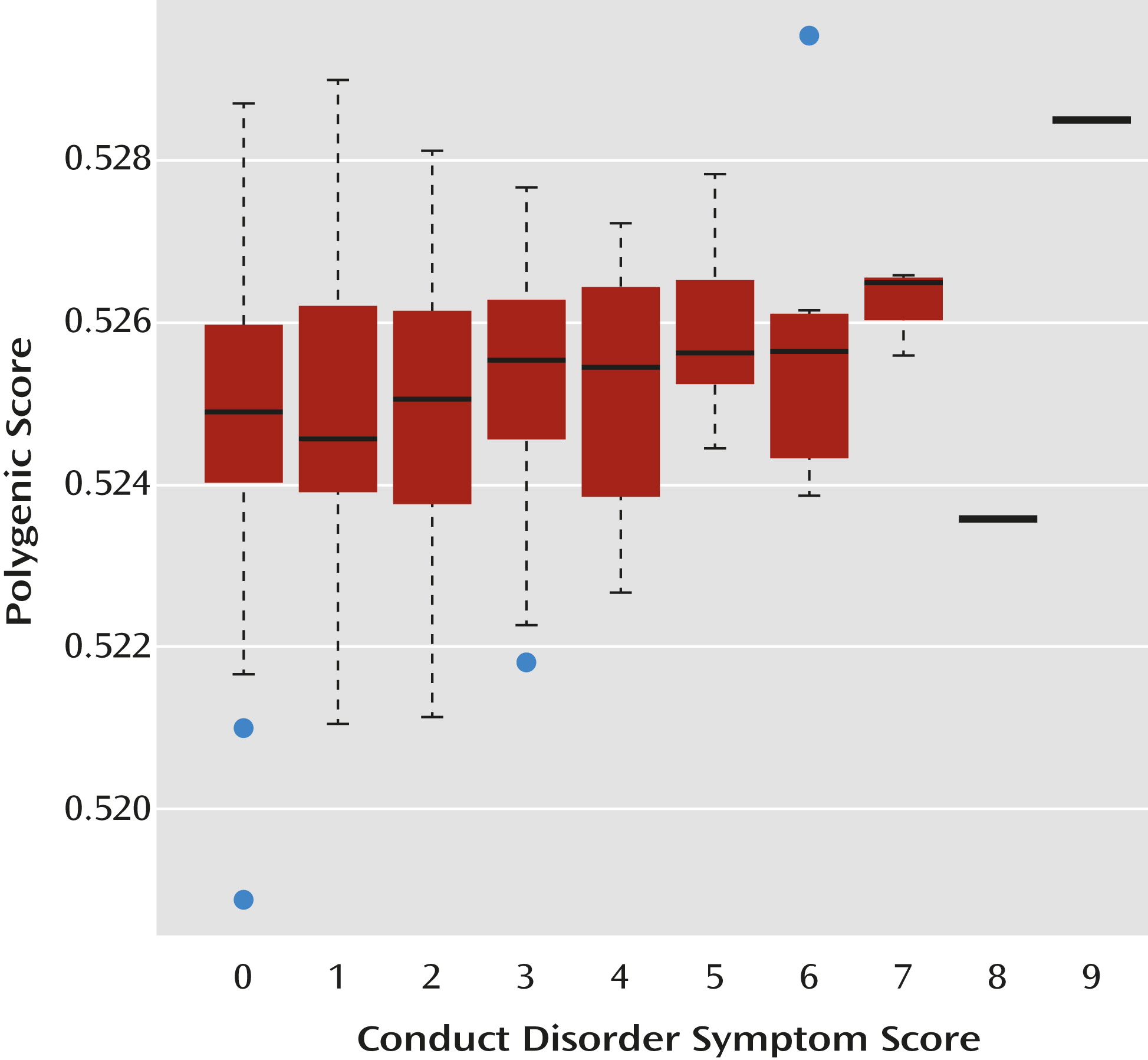
Within case subjects, ADHD polygenic risk score increased with total conduct disorder score (β=0.118, t=2.530, p=0.006). At the level of individual composite phenotypes, polygenic risk score also increased with number of aggressive conduct disorder symptoms (β=0.151, t=3.152, p=0.002), but not covert conduct disorder symptoms. These associations remained significant when controlling for sex. The distribution of risk scores showed increasing polygenic score with respect to increasing total conduct disorder scores (
Figure 1).
Discussion
We initiated this study to investigate the contribution of common genetic variants to ADHD and to test whether comorbid conduct disorder, defined categorically and dimensionally, indexed greater genetic risk at a molecular level. Our data support a polygenic component to ADHD, in that the risk score was higher in our independent sample of ADHD case subjects than in the comparison sample. To our knowledge, this is the first report to suggest that the previously published ADHD GWAS meta-analysis (
8) harbors common risk alleles that show contribution to ADHD when they are considered en masse. More importantly, as hypothesized on the basis of previous family and twin studies suggesting that comorbid conduct disorder indexes higher familial and genetic loading in ADHD, we found that ADHD risk score is particularly elevated in those with ADHD and conduct disorder compared with those who have ADHD only.
A within-case analysis of conduct disorder symptoms as a dimension rather than a category revealed similar findings, with a positive linear relationship between ADHD polygenic scores and comorbid conduct disorder symptoms. Interestingly, this association was related to aggressive, rather than covert, conduct disorder symptoms. Twin studies also suggest that these different symptom dimensions may be distinct in their genetic etiology, with stronger genetic loading for overt aggressive symptoms (
32,
37).
Overall, our study confirms the hypothesis that common genetic variants are relevant to ADHD risk. Our findings also highlight the fact that comorbid conduct disorder indexes heterogeneity in terms of genetic loading at a molecular level. Our finding that individual symptom groups of total conduct disorder scores and aggressive conduct disorder scores are significantly associated with polygenic score further underscore the point that specific clinical phenotypes can index differential genetic loading. Most of the evidence to date suggests that conduct problems index ADHD cases that are quantitatively rather than qualitatively different from the remaining ADHD cases (
38) in terms of the patterns of association with clinical, cognitive, genetic, and environmental correlates. This is in keeping with the approach taken by ICD-10, in which hyperkinetic conduct disorder is considered a subtype of ADHD/hyperkinetic disorder (
39). However, some associated factors also appear to be unique to conduct disorder in ADHD. Notably, the functional
COMT Val158Met variant has been found to be associated with conduct problems in ADHD (
40), a finding that has been replicated in six independent samples (
32,
41–
43), while meta-analysis shows that this variant is not associated with ADHD risk (
44). A separate issue of interest would be to investigate the independent contribution of genetic liability associated with conduct disorder (regardless of ADHD). There is evidence to suggest shared liabilities, but as yet, large-scale conduct disorder GWAS data sets are unavailable.
We also incidentally found that female case subjects had significantly higher ADHD risk scores than males. This finding requires replication, but it is intriguing, as it supports the hypothesis that ADHD is less common in females because a more extreme genetic load is required for the liability threshold to be surpassed. This would predict that relatives of female ADHD probands have a greater risk for ADHD than relatives of male ADHD probands, although evidence here is lacking (
3). However, there is a possible effect of ascertainment bias. Females with ADHD may be less likely to be diagnosed (or referred for diagnosis) than males with comparable severity of disorder (
45), and therefore our findings might simply reflect a higher threshold of severity for females to be diagnosed and ascertained. However, females in our sample did not have significantly more ADHD or conduct disorder symptoms than males.
As expected, ADHD severity (i.e., total ADHD score) showed association with conduct disorder scores, but the genetic findings were driven by conduct disorder, as there was no association between ADHD severity and polygenic risk. Given that all clinical case subjects have high ADHD scores by definition, and thus variance is limited, this may not be surprising.
It is noteworthy that case-control comparisons were significant for the total ADHD group and for the ADHD with conduct disorder group but did not achieve significance in the ADHD without conduct disorder group. This result might simply reflect sample size, whereby larger samples would be needed to demonstrate statistically significant differences in groups with lower genetic load (those with ADHD without conduct disorder) than in those with higher genetic load (those with ADHD with conduct disorder).
This study used a well-characterized sample of children who underwent careful phenotyping. The diagnosis of ADHD was confirmed using a semistructured research diagnostic interview, which also allowed the collection of detailed information on conduct disorder symptoms, and the measures showed high reliability. It is not possible to obtain clinical data on the comparison subjects, although failure to exclude comparison subjects with ADHD or conduct disorder would reduce our study’s statistical power, not generate false positives. One limitation of case-control and within-case studies is the possibility of population stratification. However, we limited the impact of stratification by the well-accepted approach of including derived principal components that allow for population variation (
35,
36) in our ADHD GWAS and by using hypothesis-driven analyses. Furthermore, for stratification to be an issue, it would have to be refractory to inclusion of the principal components, and the same uncorrected ethnic variation would have to be overrepresented in case subjects in both the Cardiff sample and the samples used in the meta-analysis (
46); it would also have to be specifically overrepresented in subjects with comorbid conduct disorder compared with the full ADHD sample.
The magnitudes of the polygenic effect (as defined by R
2) are small, as is typical when this method is used. The magnitudes are also smaller than some of those published for schizophrenia (R
2=3%–6% [
24,
25]) but not all (
47). This method is also very sensitive to sample size, and available GWAS data sets for ADHD are very much smaller than those for schizophrenia. In the most recent schizophrenia analysis, with more than 9,000 case subjects and 12,000 comparison individuals (
24), the total amount of variance explained was estimated to be 6%, whereas in an earlier analysis of 3,000 case subjects and 3,000 comparison subjects by the International Schizophrenia Consortium (
25), the estimate was 3%. Our estimate that 0.1% of the variance explained is based on much smaller samples, with 452 ADHD case subjects and 5,081 comparison subjects. Another factor that would reduce explained variance is heterogeneity across samples. This could plausibly be higher for some disorders than others—for example, through international differences in clinical service provision and thus in ascertainment, as well as other variability, such as ethnic composition. It is also important to note that GWAS SNP arrays do not completely capture relevant genetic variation; they “tag” potentially causal variants, and the arrays do not capture the full spectrum of allelic frequencies or allele types (e.g., repeat sequence polymorphisms such as variable number tandem repeats). Overall, we expect that as more ADHD and comparison samples with more comprehensive genetic capture become available, more variance will be explained. Finally, we also acknowledge the contribution of environmental risk factors and gene-environment interplay, although this was not the focus of the present analyses.
These findings suggest that ADHD, like other psychiatric disorders, can be considered a polygenic disorder, the architecture of which includes common as well as rare alleles (
10). They are also compatible with our earlier work showing overlap between the biological processes enriched for weak ADHD SNP association signals and those enriched for rare copy number variants (
5). Given the evidence for contribution of common variants, the future acquisition and genetic analysis of much larger ADHD samples in an attempt to capture relevant genetic variation is crucial. The aim is not to simply identify single “significant” SNPs of small effect size, but rather to utilize the spectrum of associated common and rare genetic risk variants to uncover novel clues for risk mechanisms and underlying biology and to inform our conceptualization of ADHD.
In summary, we found that common genetic variation appears relevant to ADHD and that a higher loading for common ADHD genetic risk variants is indexed by comorbid conduct disorder, especially aggressive symptoms. Our results also suggest that the previously published ADHD GWAS meta-analysis contains weak but true associations to common variants, support for which falls below currently accepted genome-wide significance levels. The findings highlight the fact that aggression in ADHD, as an index of clinical severity, is underpinned by higher genetic loading at a molecular level. They also illustrate that for hypothesis-driven research, careful phenotyping is still useful in psychiatric genetic studies.
Acknowledgments
Supported by Wellcome Trust, the Medical Research Council (U.K.), and Action Research. Acknowledgments for the published ADHD meta-analysis data are detailed in reference 1.
The authors acknowledge the members of the Psychiatric Genomics Consortium, ADHD Subgroup, for providing data and analyses for the ADHD meta-analysis used as the discovery sample in this manuscript: Erik Willcutt, Mark Daly, Thuy Trang Nguyen, Susanne Walitza, Helmut Schäfer, Michael Gill, Joseph Sergeant, Eric Mick, Susan Smalley, Sandra Loo, Josephine Elia, Alexandre Todorov, Ana Miranda, Fernando Mulas, Richard Ebstein, and Stan Nelson.