Suicide is considered a major preventable public health problem and is the 10th leading cause of death in the United States for all age groups combined. It is generally accepted that suicide marks the tragic endpoint of a highly complex behavior shaped by social, developmental, and neurobiological factors that translate into a predisposition for the act itself. For example, altered serotonergic activity in the prefrontal cortex, which appears to be partially independent of the changes observed in major depression, is thought to play an important role (
1,
2). Furthermore, the concordance rate for serious suicide attempts in monozygotic twins has been reported to be up to 17-fold greater than in dizygotic twins, and heritability estimates for serious suicide attempts are estimated to be as high as 55% (
2). On the other hand, recent genome-wide association studies of nearly 9,000 individuals diagnosed with a mood disorder, approximately one-third of whom had a history of attempted suicide, effectively ruled out common genetic variants and polymorphisms of large effect (
3). Following this approach, much larger cohorts will be required in order to gain insight into what appears to be a highly polygenic risk architecture.
In this issue, Labonté et al. (
4) take a different approach by focusing on genome-wide promoter mapping of DNA cytosine methylation as an epigenetic marking that decorates the human genome and is thought to play a critical role in the regulation of gene expression. Promoters are regulatory DNA sequences adjacent to the start site of a gene, regulating its expression. DNA methylation in promoter sequences is often associated with a repressive chromatin state and lower gene expression activity. Methylation of DNA occurs for different reasons, but life experiences, generally stressful ones, are often associated with changes in methylation of an animal’s DNA. Labonté et al. explored DNA methylation changes both in neuronal and nonneuronal chromatin from postmortem hippocampus of 46 individuals who died by suicide and 16 comparison subjects from the Quebec Suicide Brain Bank. Because DNA cytosine methylation and other epigenetic modifications such as histone acetylation are understood as a molecular bridge linking gene expression in brain cells to environment, exposure, and experience, methylation stands at the forefront of mechanisms waiting to be explored to better understand psychiatric maladies such as depression and suicide that are not rooted solely in faulty genetics in the overwhelming majority of cases.
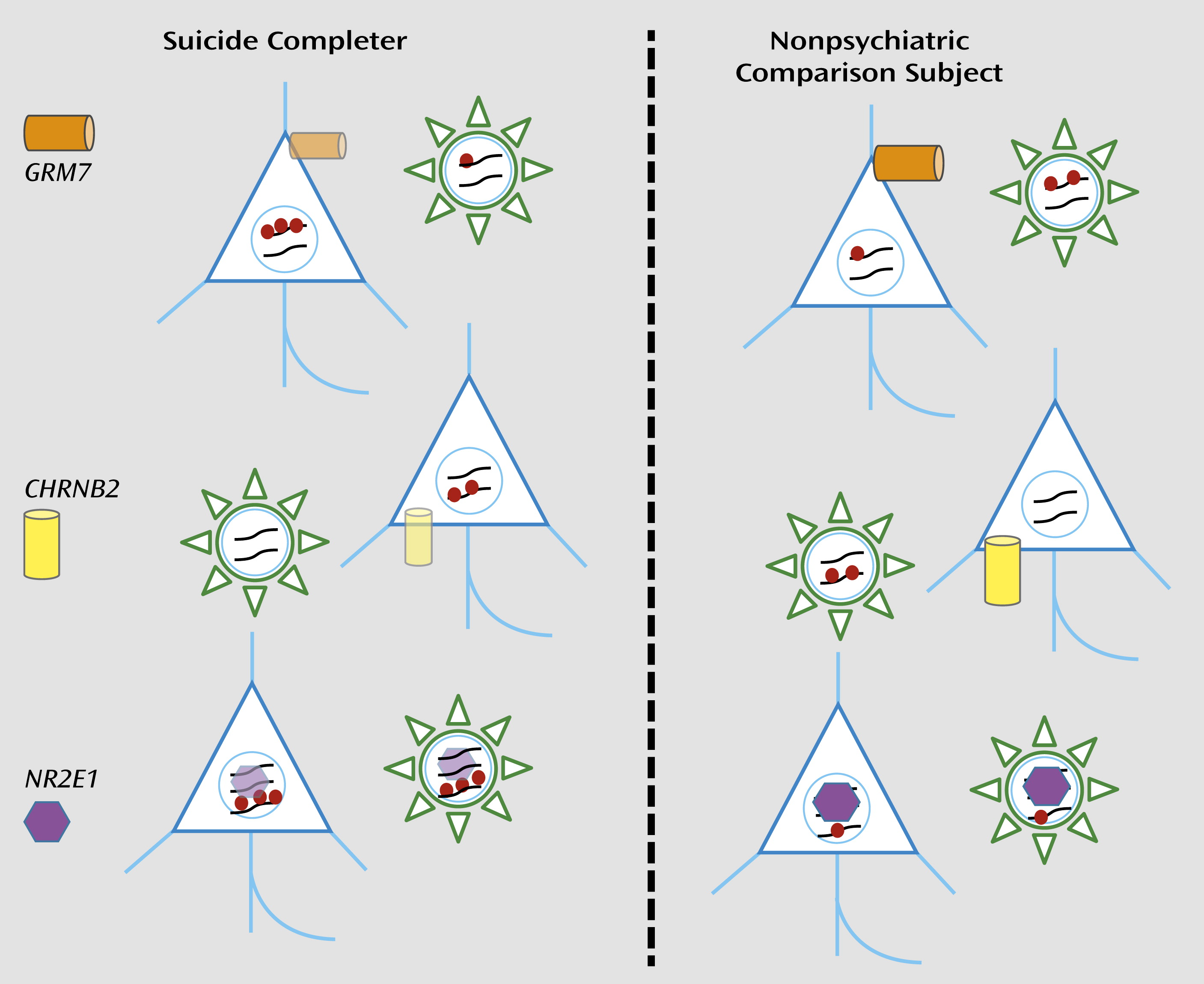
Labonté et al. found that hippocampal DNA from suicide completers showed DNA methylation changes at more than 300 gene loci. Interestingly, this pool of epigenetically altered gene promoters was significantly enriched for genes implicated in cognition, learning, and memory as well as synaptic transmission. In addition, many of the observed DNA methylation changes were specific for the hippocampal neurons and were not observed in glial or other nonneuronal DNA from the same tissue (
Figure 1). Therefore, the study findings underscore the fact that suicide and depression are, to a certain degree, disorders of the neuronal genome. From a broader perspective, this study, along with similar work on other psychiatric conditions (
5), further emphasizes the fact that genome exploration in brain tissue is ideally done in a cell type-specific fashion, in order to fully capture disease-relevant epigenetic signals that may affect only a subtype of neuron or glia.
The list of genes subject to promoter DNA hypermethylation and decreased hippocampal expression in suicide completers included metabotropic glutamate receptors and nicotinic acetylcholine receptor subunits, and at least one gene,
NR2E1, encoding a nuclear receptor related to steroid hormone sensors that when mutated in mice results in defective development and hyperaggressive and violent behaviors in both sexes. Tellingly, this mutant mouse line has been named
Fierce (
6). It is encouraging that a subset of epigenetically altered genes in the brains of suicide completers, including
NR2E1, elicit a congruent behavioral phenotype in a mouse model. Whether this finding will open opportunities to explore radically novel treatment options, or at least increase insight into disease-relevant molecular mechanisms in a preclinical model system, remains unclear. Obviously, suicidal behavior is difficult to model in the animal.
Looking forward, are Labonté et al. (
4) pointing us toward an epigenetic risk architecture that reflects a predisposition to suicide, by reporting a large (but by no means infinite) number—300 to 400—of genomic loci? The answer may not be straightforward, because recent work from the same group, using similar approaches in a different sample of suicide completers who had experienced severe abuse in early childhood, yielded again hundreds of epigenetically altered loci (
7). However, the overlap of specific loci between these two studies is limited (
4,
7), which led the authors to the conclusion that DNA methylation changes in the brains of suicide completers who had suffered childhood abuse may differ from those observed in suicide completers who presumably did not experience early-life adversity (
4). Interestingly, the hippocampal DNA methylation studies (
4,
7) both reported that epigenetic changes are more prominent in the neuronal as compared with the nonneuronal fraction. Together with a recent transcriptome study of prefrontal specimens from a Hungarian brain collection in which disruption of gene expression networks important for neuronal communication and synaptic signaling was reported (
8), these findings contribute to converging evidence that suicide and depression are associated with widespread changes in the molecular architecture of nerve cells residing in key structures of the corticolimbic circuitry.
It will also be important to compare these transcriptome and DNA methylome results from postmortem brain studies with similar data sets obtained from blood samples of individuals with a history of suicide attempts (
9). Moreover, most epigenetic markings, including DNA methylation, are now considered to undergo dynamic and reversible changes during life, for example, in response to acute stressors. Therefore, longitudinal studies of tissue or cells easily accessible from living subjects who are able to give consent, complemented by animal work, will be important in order to distinguish between epigenetic changes reflecting an enduring trait triggered by a single early significant stressor or other factors operating earlier in life, as opposed to an acute process reflecting recent or current life events. With the emerging evidence that many neuronal gene promoters are epigenetically dysregulated, novel drugs targeting chromatin-associated proteins surely represent the next frontier for preclinical research in this area (
10).