Genetic variation at the ABCB1 (MDR1) locus, which encodes P-glycoprotein, has been studied as a predictor of treatment outcomes for several medications (
5,
6). The primary hypothesis has been that genetic variants affecting P-glycoprotein abundance or function could alter brain concentrations of substrate medications. Substrate brain antidepressant levels have been shown to be higher in mice lacking P-glycoprotein function than in mice with normal P-glycoprotein function (
4). Several ABCB1 single-nucleotide polymorphisms (SNPs) may be clinical predictors of antidepressant efficacy (
7–
10) or side effects (
11), but results have not been consistent (
12). Some studies have shown no predictive value for ABCB1 SNPs, particularly earlier studies that explored exonic alleles in medical disorders (
13–
17). These varying accounts may be explained by clinical heterogeneity among patient samples (e.g., inpatients versus outpatients), differing interactions of substrate medications with P-glycoprotein, specific alleles explored, and sample size. Also, ABCB1 genotypes have not been assessed in large-scale prospective clinical trials in which DNA was collected before treatment.
Cognitive impairment is common in patients with major depressive disorder (
18) and may be assessed using behavioral performance tests (
19,
20,
21). In the International Study to Predict Optimized Treatment in Depression (iSPOT-D), we observed that performance on pretreatment behavioral tests of general and emotional cognition predicted posttreatment outcomes (
21,
22). However, it is unknown whether pharmacogenetic prediction can be used to improve treatment outcome in depressed patients with cognitive changes.
We examined whether 10 ABCB1 SNPs are predictors of remission and side effects in treatment with three commonly prescribed antidepressants in patients from the iSPOT-D cohort who provided DNA. We also examined the effects of ABCB1 SNPs in patients with intact and impaired cognition.
Method
Overview
iSPOT-D is a multiple-phase, multisite randomized controlled trial that explores biomarker predictors of outcomes in 1,008 participants with major depressive disorder randomly assigned to receive escitalopram, sertraline, or extended-release venlafaxine. Details of the study design and response and remission rates for the sample have been described previously (
22,
23). In the present study, we tested whether specific SNP alleles, chosen based on associations reported in the literature, predict acute response to antidepressants and/or moderate a differential response to specific types of antidepressants. We studied 888 participants with MDR1 genotypes, of whom 683 completed at least 2 weeks of treatment (the modified intent-to-treat sample), 84% of whom (N=576) completed the full 8 weeks of treatment (the per-protocol sample) (see Figure S1 in the
data supplement that accompanies the online edition of this article). Given the potential bias from the per-protocol sample, we focused on the modified intent-to-treat sample and then tested whether results were consistent in the per-protocol sample. Participants were recruited from eight academic and nine private sites in five countries (mean enrollment per site, N=56, SD=96) and had ABCB1 genotypes and cognitive testing at pretreatment baseline.
Study Participants
The study enrolled adults (18–65 years old) who had a diagnosis of current, single-episode, or recurrent nonpsychotic major depressive disorder and whose treating clinician approved treatment with an antidepressant drug. See Figure S2 in the
data supplement for inclusion and exclusion criteria (
22). Psychiatric inclusion and exclusion criteria were based on the Mini International Neuropsychiatric Interview (
24), the 17-item Hamilton Depression Rating Scale (HAM-D) (
25) to assess severity (a score ≥16 was required for inclusion), and a urine toxicology screen. The Mini International Neuropsychiatric Interview was also used to assess comorbid psychiatric disorders. Race and ethnicity were assessed by self-report as part of a computerized questionnaire using standard World Health Organization formats (
Table 1).
Written informed consent was obtained from all participants after the procedures had been fully explained. The study was approved by institutional or ethical review boards at each site, and its protocols were in compliance with International Conference on Harmonization and Good Clinical Practice principles, the U.S. Food and Drug Administration Code of Federal Regulations, and country-specific guidelines.
Study Treatments
Participants were randomly assigned to receive escitalopram, sertraline, or extended-release venlafaxine. Randomization was undertaken centrally using Phase Forward’s validated web-based Interactive Response Technology application to implement a blocked randomization procedure (block size of 12, across sites). Investigators, raters, and participants were not blind to treatment assignment. Medications were prescribed and dosages adjusted by treating clinicians according to routine clinical practice, but following the recommended dosage ranges. Psychotropic medication was discontinued prior to randomization except for occasional (i.e., ≤1 dose/week) use of sleep aids, anxiolytics, and medications to manage antidepressant-induced side effects (e.g., nausea), which are commonly used in practice. Treatments for concurrent general medical conditions were allowed and recorded.
Outcome Measures
We focused on two planned outcome measures: remission, defined as a score ≤5 on the 16-item Quick Inventory of Depressive Symptomatology (QIDS-SR) (
26), and the sum of the three items of the Frequency, Intensity, and Burden of Side Effect Rating (FIBSER) scale (
27) (referred to here as the FIBSER sum score). The QIDS-SR and the FIBSER were the only outcome measures to be collected at baseline, at the 8-week posttreatment follow-up, and during treatment-phase telephone monitoring sessions (at weeks 1, 2, 4, and 6).
Behavioral Tests of General and Emotional Cognition
At baseline, participants completed a computerized test battery that evaluated 13 cognitive and emotional capacities, including response speed, decision speed, processing speed, attention/working memory, cognitive flexibility, response inhibition, verbal memory, executive function, emotion identification, and the implicit priming of simple decision by emotion (
28,
29). To create summary measures of each test, we followed our previously established procedure for normalizing each measure to the benchmark from 336 healthy controls (i.e., as standardized z-scores relative to a control mean of 0) and averaged normalized measures (e.g., accuracy and reaction time) within each test. Values on each measure were aligned such that positive indicated better performance and negative indicated worse performance.
Heterogeneity in Cognitive and Emotional Test Performance
Consistent with our previous report (
21), participants fell into two clusters of cognitive and emotional test performance: an “intact” cluster who performed on average within the healthy range of z>−0.5, and an “impaired” cluster who performed on average well below the healthy norm, at z<−0.5. This clustering method was reproducible using alternative assumption-free classification methods such as latent class analysis (
21). In the modified intent-to-treat sample, 494 participants were intact and 189 were impaired. In the per-protocol sample, 418 were intact and 158 were impaired.
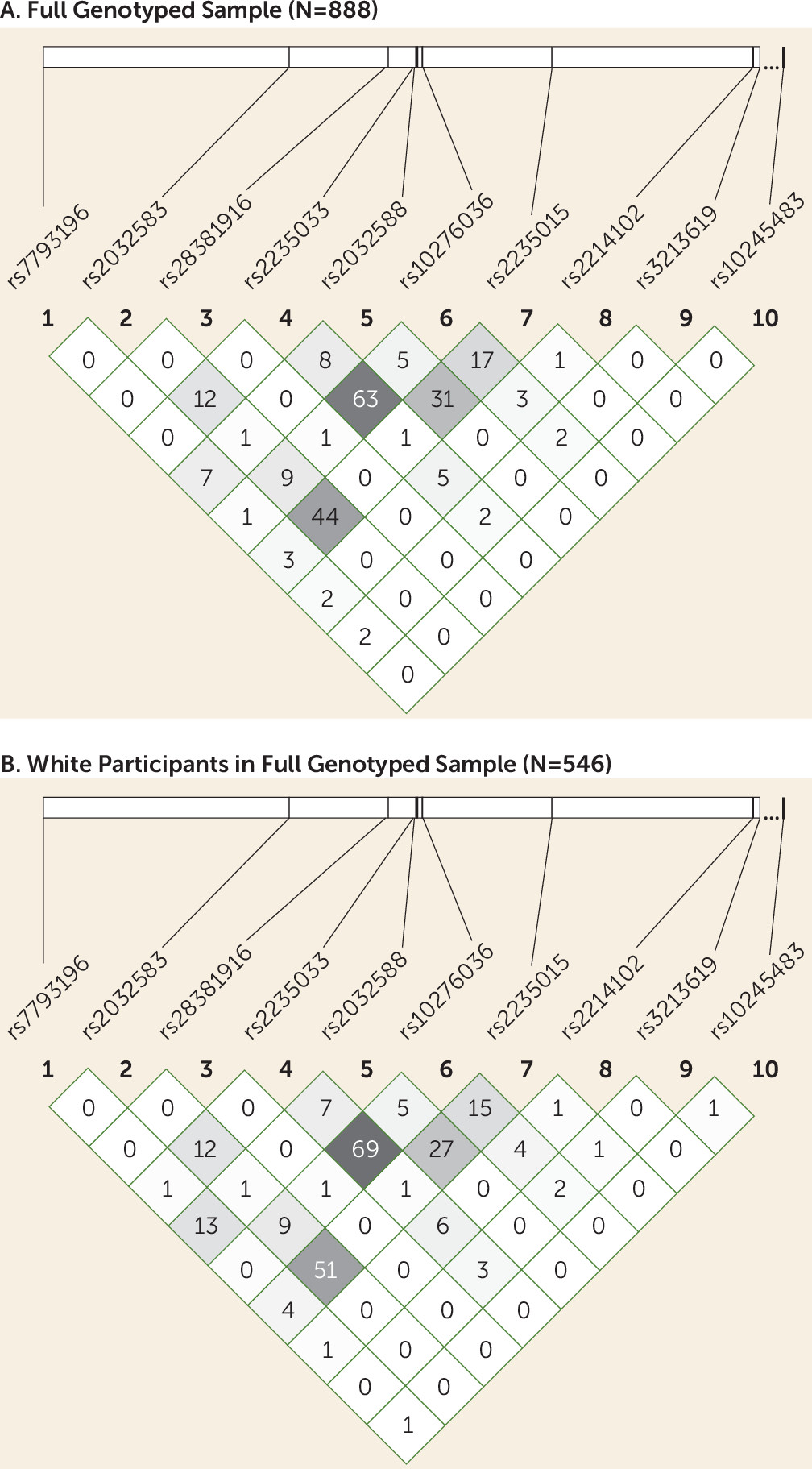
Genetic Analysis
We genotyped 10 SNPs in or near the ABCB1 gene (
Table 2), chosen based on previously reported associations with ABCB1 function or on pharmacogenetic studies of ABCB1 substrate drug treatment outcomes (
7–
9,
11,
13,
30–
33). Four of these have been investigated for predicting antidepressant response or remission: rs2032583 (
7,
8,
11,
34,
35), rs2235033 (
8), rs2235015 (
7,
8,
11), and rs10245483 (
7). Others have been studied in other diseases (e.g., rs10276036, rs2214102, and rs32313619 in cancer risk) (
36,
37).
DNA extraction was performed from EDTA-treated blood using the Puregene DNA method (Qiagen, Valencia, Calif.). Genotyping was performed using the Illumina VeraCode Golden Gate SNP genotyping platform (Illumina, Hayward, Calif.) by Covance, Inc. (Seattle). To screen for genotyping errors, we checked for deviation from Hardy-Weinberg equilibrium (
38). Because the sample was ethnically heterogeneous, and population stratification can result in deviation from Hardy-Weinberg equilibrium, we also tested for Hardy-Weinberg equilibrium in participants with self-reported white ethnicity. Haploview, version 4.2 (
39), was used to calculate p values for Hardy-Weinberg equilibrium deviation and to calculate linkage disequilibrium among SNPs.
Statistical Analysis
Logistic regression analyses were performed with remission (defined by QIDS-SR score) as the binary dependent outcome measure. We tested for effects of individual ABCB1 SNPs and the effects of SNP-by-treatment interactions on remission. Thus, predictors in the model were ABCB1 SNPs entered simultaneously (and with each SNP coded by number of minor alleles as 0, 1, and 2 using an additive allelic model), treatment defined as a categorical variable, and the interaction of these predictors. We did not include rs3213619 in analyses because of the absence of minor allele carriers for this SNP, and thus the analyses were undertaken with nine SNPs in total. Site was included as a covariate in the regression model. Potential confounders included pretreatment symptom severity, age, and treatment duration. When one or more of these variables contributed to remission status, we included them in the regression model as additional covariates. Within the overall regression model, the Wald statistic (W) was used to assess the significance of the contribution of each predictor. To account for the testing of multiple SNPs, SNP p values of 0.05/18 ≤0.0028 were considered significant using a Bonferroni correction for nine SNPs, each tested for one main effect and one interaction effect. All p values <0.05 are reported for completedness, because of the possibility of false negative results at this significance level. Based on the exponential beta values in the regression models, we generated the odds ratio for each significant effect. The odds ratio for interaction terms reflects how much greater or lower the likelihood of remission or side effects is in a multiplicative sense between treatment arms according to number of minor alleles. To assess potential clinical applicability we computed the number needed to treat for significant SNP effects. To account for genotype frequencies, we also calculated the numbers needed to screen (
40).
Linear regression analyses were run with FIBSER sum score as the linear dependent outcome measure. Predictors were again the number of minor alleles for each SNP, treatment, and the SNP-by-treatment interaction. The t statistic was used to assess the significance of the contribution of each predictor.
In a second set of logistic (for QIDS-SR-based remission) and linear (for FIBSER sum score) regressions, cognitive status (impaired, intact) was included as an additional predictor to assess SNP-by-cognition interactions. We again tested the overall regression model, the individual contributions of SNP-by-cognition and SNP-by-treatment-by-cognition interactions, and the odds ratio for significant effects.
Our sample size was powered to achieve at least 89% power to detect even small effects for the nine SNP predictors of interest and interactions with treatment at p<0.05. This power calculation applied to the smaller per-protocol subsample from our two analytic samples. Our analytic models were designed to identify each SNP’s unique predictive contributions while taking account of the effects of other SNPs. Because genetic effects can depend on ethnic background, we examined the full sample and the subsamples of self-reported whites and nonwhites.
We compared baseline characteristics of the impaired and intact cognition groups in the modified intent-to-treat and per-protocol cohorts using t tests and chi-square tests. Similar analyses were performed comparing the demographic and clinical characteristics of the modified intent-to-treat and per-protocol samples with those patients excluded from genetic analyses.
Discussion
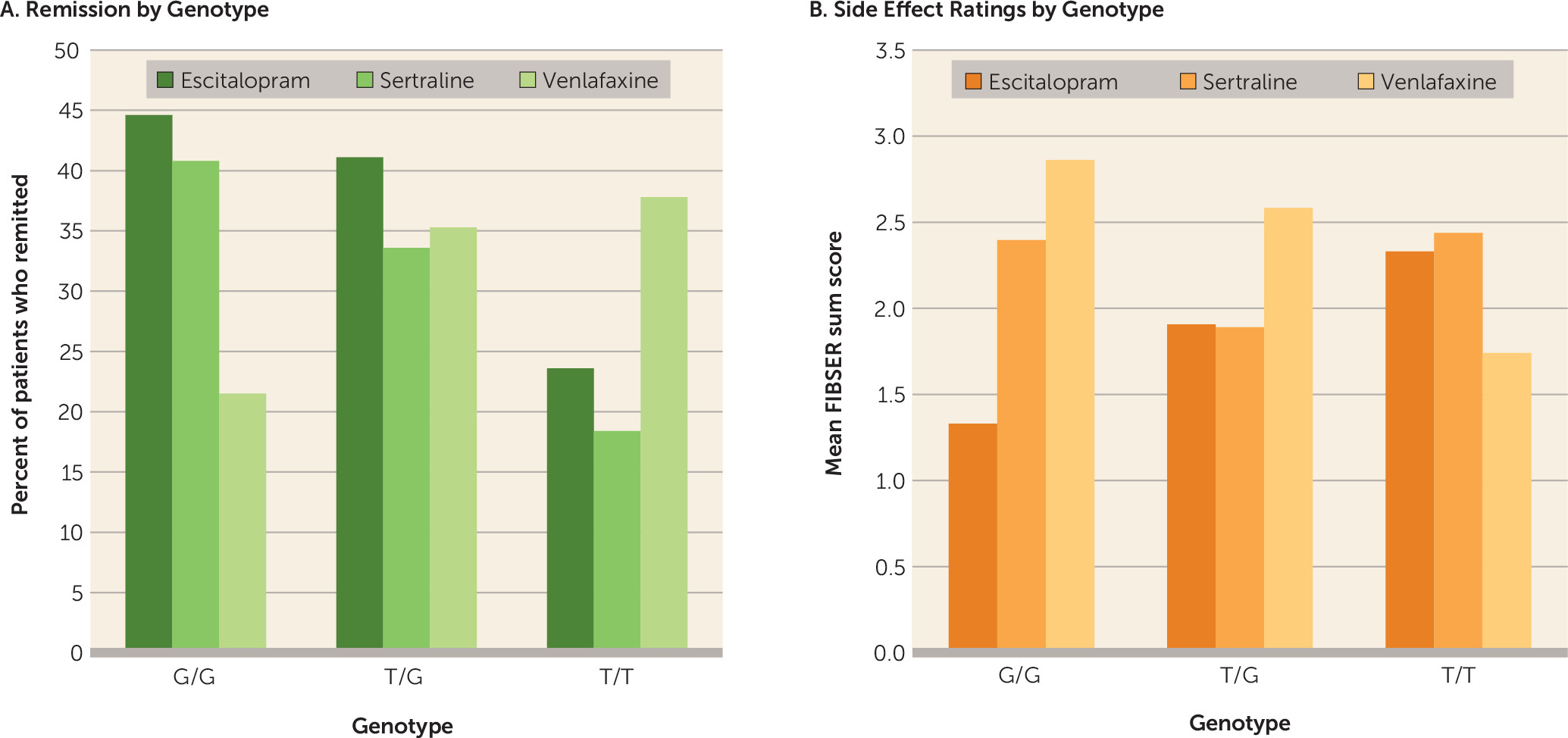
Our results demonstrate novel effects of the rs10245483 variant, which is approximately 2.5 Mb upstream of the ABCB1 locus, on antidepressant outcomes. The T minor allele has been reported to result in higher P-glycoprotein expression in a lymphoblast cell line model (
31) when found in combination with minor alleles of the rs28656907/rs28373093 dinucleotide pair. Cell line models for ABCB-1 have been shown to be have limited predictive value for in vivo activity (
41). We observed a clinical predictive effect for rs10245483 without genotyping the rs28656907/rs28373093 dinucleotide pair. Increased P-glycoprotein expression could result in enhanced clearance of antidepressants from the brain, which could explain why rs10245483 T allele homozygotes demonstrated significantly poorer responses to escitalopram and sertraline. It is not apparent why T homozygotes did well on venlafaxine. Possibly the difference in the drugs’ monoaminergic activity, with escitalopram and sertraline being selective serotonin reuptake inhibitors and venlafaxine a mixed norepinephrine-serotonin reuptake inhibitor, play a role. The association of cognitive impairment with a better response to venlafaxine in the T/T homozygotes would be in keeping with a particular need for a noradrenergic effect (i.e., the noradrenergic effect of venlafaxine may specifically improve cognition in subjects with poor cognition).
In contrast, rs10245483 had opposite actions on side effects depending on medication. Minor allele carriers who demonstrated lower efficacy with escitalopram also experienced greater side effects. We recently observed similar reciprocal prediction of efficacy versus side effects for variants of rs2235040 in a study of chronic depression (
34). MDR1 variants that predict lower antidepressant response but greater side effects could reflect nonresponding patients being treated with higher dosages that produce greater peripheral side effects resulting from higher tissue concentrations where P-glycoprotein is not expressed. In contrast, greater efficacy and lower side effects with venlafaxine were observed in minor allele carriers. Although escitalopram, sertraline, and venlafaxine are P-glycoprotein substrates (
4,
42–
44), they may differ in their relative induction versus inhibition effects on the P-glycoprotein pump, much as agents can be not only substrates but also inducers or inhibitors of P4502D6 enzymes. Sertraline inhibits P-glycoprotein activity at the blood-brain barrier (
45), such that chronic treatment increases sertraline brain levels. This could offset the minor allele increased efflux effect. Escitalopram is neutral in its activity. Venlafaxine induces P-glycoprotein expression (
46), and chronic treatment could induce P-glycoprotein and lower brain (and perhaps serum) concentrations. However, at the dosages used in our study, P-glycoprotein induction by venlafaxine may be negligible (
46). We did find that among venlafaxine-treated participants, rs10245483 had a significant effect on side effects, but whether this is related to the P-glycoprotein-inducing properties of the drug remains unknown. Further study, using measures of central drug concentration, efficacy, and side effects, is required to better understand the differences in genetic prediction among the three agents.
Little else is known about the clinical effects of rs10245483. Our study in an elderly depressed sample (
7) found no evidence of an effect of rs10245483 on paroxetine or mirtazapine efficacy. Other ABCB1 antidepressant pharmacogenetic studies have not assessed this variant (
8,
11,
13,
32).
The minor C allele for the intronic SNP rs2032583 has been associated with shorter time to remission on substrate medications (
7,
8) but greater antidepressant side effects (
11). However, we recently reported it to be associated with greater remission and lower side effect burden in a chronic depression cohort treated primarily with escitalopram or sertraline (
34,
35), although that finding did not withstand correction for multiple testing. In the present study, we found no evidence for a significant effect of this SNP on remission. We did note significant prediction of response for the minor allele (data not shown). Given these findings, rs2032583 seems a less robust predictor of outcome.
SNP rs2235033 has been associated with likelihood of remission (
8), a finding not replicated in the present sample. Similarly, rs2235015 has been shown to predict remission (
8), but it was not predictive in this sample, which is in agreement with other studies (
7,
11).
In the full cohort of iSPOT-D completers, we recently reported that impairment on tests of cognitive and emotional function predicted poorer antidepressant outcomes (
21). Patients with cognitive impairment and major depression may represent a distinct population with unique clinical characteristics and possibly a different response to treatment (
18). For example, patients with depression and cognitive impairment tend to be older and have a longer duration of illness, as we found in the present sample; however, our sample was by design not elderly, and the effect of age on remission was included in the analyses.
Another explanation is that the cognitively impaired nonelderly participants represent what is classically referred to as suffering from a significant depression, in contrast to patients with milder depression who often barely meet criteria for major depressive disorder and are often recruited into clinical trials. Thus, the cognitively impaired may represent a distinct endophenotype for depression defined by these impairments and associated disruptions to cognitive brain circuitry. We are following up our studies on cognition with other biomarkers to better understand the neurobiology of these patients.
From a pharmacogenetic perspective, it may be that because of age-associated changes in impaired patients, differences in brain concentrations due to altered P-glycoprotein function have a more pronounced effect in older than in younger individuals. Certainly, greater sensitivity to a variety of medications has been associated with aging (
47,
48), and the function of the P-glycoprotein pump declines with age (
49). In the present study, there was a strong overall association between age and remission, with older patients showing less frequent response than younger ones. Even after controlling for age, we observed a significant interaction of cognition and rs10245483 on treatment response.
Our study had several limitations that must be considered. We only included patients who completed at least 2 weeks of treatment. There could be pharmacogenetic effects that affected those who dropped out early. ABCB1 is a large gene, and we assessed a limited number of SNPs. There could be other ABCB1 pharmacogenetic markers that we did not test, in particular the rs28656907/rs28373093 dinucleotide pair that modulates the effect of rs10245483. Although dosages were similar across cohorts and genotypes, we did not determine serum antidepressant concentrations, which could affect outcomes. Because iSPOT-D is a real-world outcomes study that includes primary care providers, the dosages used were somewhat lower than those used in traditional clinical drug trials. Higher dosages might have resulted in different pharmacogenetic effects.
In conclusion, we found that the functional SNP rs10245483, located upstream from the ABCB1 gene, affects antidepressant medication efficacy and side effects. The effect depended on the specific medication. Our results suggest that ABCB1 variation is drug specific and that further study is needed to understand the differences among the agents. The results also suggest that other pharmacogenetic cohorts should be examined for effects in cognitively impaired subgroups.