Although a broad-based symptom control is key to the effective treatment of schizophrenia, second-generation antipsychotics act mainly on positive symptoms (
1). Moreover, second-generation antipsychotics can cause various adverse effects, which may worsen the patient’s ability to function, diminish subjective well-being and quality of life, and contribute to poor treatment adherence (
2). Long-term antipsychotic medication adherence is crucial to preventing relapse (
3). Different second-generation antipsychotics have distinct adverse effect profiles (
4,
5), likely influenced by their varying pharmacological profiles (
6). For example, clozapine and olanzapine are most often associated with weight gain and related metabolic abnormalities (
5); clozapine, olanzapine, quetiapine, and ziprasidone may cause sedation (
4); ziprasidone is associated with QTc interval prolongation (
4); aripiprazole is associated with activating adverse effects, including restlessness/akathisia (
7); lurasidone, paliperidone, and risperidone have been associated with extrapyramidal symptoms; and paliperidone and risperidone have been linked to an increase in prolactin and potentially resultant sexual adverse effects (
4). Selecting a second-generation antipsychotic requires consideration of the balance between activating and sedating adverse effects, as well as attention to extrapyramidal symptoms and cardiovascular risk. New treatments that are better tolerated are needed to optimize physical health as well as social functioning.
Brexpiprazole is a serotonin-dopamine activity modulator that acts as a partial agonist at serotonin 5-HT
1A and dopamine D
2 receptors at similar potencies and as an antagonist at 5-HT
2A and noradrenaline alpha
1B/2C receptors (
8). Preclinical models have demonstrated brexpiprazole’s efficacious functional D
2 receptor partial agonist activity and antipsychotic-like profile (
9). Brexpiprazole shows partial agonism with lower intrinsic activity at the D
2 receptor and stronger antagonism at the 5-HT
2A receptor than the only currently available D
2 partial agonist, aripiprazole (
8), suggesting a relatively lower potential to induce D
2 partial agonist-mediated adverse effects, e.g., akathisia, insomnia, restlessness, and nausea (
10). The potential to induce D
2 antagonist-like adverse effects, e.g., extrapyramidal symptoms, hyperprolactinemia, and possibly tardive dyskinesia, is also considered to be lower than with full D
2 antagonists (
8). Further, brexpiprazole’s balanced 5-HT
2A/D
2 and 5-HT
1A/D
2 receptor binding profile may contribute to low incidences of both activating and neuromotor adverse effects clinically (
8). Finally, brexpiprazole has a moderate affinity, relative to D
2/5-HT
1A receptor affinity, for histamine H
1 receptors (
8), which may result in low sedation levels.
The objective of this study was to evaluate the efficacy, safety, and tolerability of three fixed dosages of brexpiprazole (0.25, 2, and 4 mg/day) compared with placebo in adults with acute exacerbation of schizophrenia. We hypothesized that brexpiprazole at 2 and 4 mg would be more efficacious than placebo and well tolerated; the 0.25-mg dosage was hypothesized to be ineffective on the basis of preclinical data.
Method
Patients
Patients were recruited at 65 study centers in the United States (35.8% of randomly assigned patients), Ukraine (18.1%), Romania (17.1%), Serbia (11.8%), Latvia (4.9%), Malaysia (3.3%), Japan (3.0%), Poland (2.5%), South Korea (2.4%), and Canada (1.1%). Eligible patients were 18–65 years old, had a DSM-IV-TR diagnosis of schizophrenia confirmed by the Mini International Neuropsychiatric Interview for Schizophrenia and Psychotic Disorders Studies (
11), experienced an acute exacerbation, and would benefit from hospitalization or continued hospitalization for treatment. Patients were excluded if they had a first episode of schizophrenia, a DSM-IV-TR axis I diagnosis other than schizophrenia, clinically significant tardive dyskinesia, substance abuse or dependence in the previous 180 days, or a clinically significant medical condition.
Study Design
This randomized, double-blind, placebo-controlled phase 3 study (ClinicalTrials.gov identifier: NCT01396421; the VECTOR trial) was conducted from August 2011 to December 2013. The study comprised a 14-day screening phase, a 6-week double-blind treatment phase, and a 30-day follow-up phase, as shown in Figure S1 in the data supplement accompanying the online version of this article.
Eligible patients were randomly assigned by using an interactive voice or web-based response system to 0.25, 2, or 4 mg of oral brexpiprazole or placebo once daily (1:2:2:2). Blocks of randomization numbers based on a computer-generated permuted-block randomization schedule were assigned to each study center. In the groups receiving 2 or 4 mg of brexpiprazole, dosing began at 1 mg/day and was titrated to 2 mg on day 5 and 4 mg on day 8.
Assessments
Efficacy was assessed by using the Positive and Negative Syndrome Scale (PANSS) (
12), the Clinical Global Impressions Scale (CGI) severity of illness and improvement scales (
13, pp. 218–222), and the Personal and Social Performance scale (
14,
15). The PANSS and CGI scales were completed at screening and baseline and at weekly intervals during treatment; the Personal and Social Performance scale was administered at the baseline, week 3, and week 6 visits.
The primary efficacy measure was change from baseline at week 6 in PANSS total score. Secondary efficacy measures were change from baseline at weeks 1–5 in PANSS total score; change from baseline at week 6 in the CGI severity rating (key secondary endpoint measure), Personal and Social Performance score, and PANSS positive and negative symptom subscale scores; CGI improvement rating at week 6; rate of response at week 6 (defined as change from baseline ≥30% in PANSS total score or CGI improvement score of 1 or 2); discontinuation rate due to lack of efficacy; and change from baseline at week 6 in PANSS score on the excited component (comprising excitement, hostility, tension, uncooperativeness, and poor impulse control scores) (
16) and in scores on the five PANSS factors defined by Marder et al. (
17). Additional post hoc exploratory analyses regarding different response definitions were conducted, based on reductions of ≥20%, ≥40%, and ≥50% in PANSS total score.
Safety and tolerability variables were adverse events, body weight, laboratory measurements, vital signs, electrocardiogram, Barnes Akathisia Rating Scale (
18), Simpson Angus Scale (
19) (the Drug-Induced Extrapyramidal Symptom Scale [
20] was used in Japan), Abnormal Involuntary Movement Scale (AIMS) (
13, pp. 534–537), and Columbia-Suicide Severity Rating Scale (
21,
22).
Data Analysis
Sample size calculations were based on a predicted difference for 2 and 4 mg of brexpiprazole versus placebo of 7.5 points (SD=20) on the PANSS total score. A total sample size of 630 evaluable patients was projected to yield ≥90% power to detect treatment effects at a two-tailed alpha of 0.025.
The safety population included all randomly assigned patients taking at least one dose of study medication; the efficacy population included only patients with efficacy evaluations at baseline and on at least one occasion after baseline. The primary efficacy endpoint measure was analyzed by using a mixed model for repeated measures. The model included fixed-effect factors of treatment, site, visit, treatment–visit interaction, and fixed-effect covariates of baseline value and baseline–visit interaction. A two-step testing approach was used to control the family-wise error rate of multiple comparisons (
23). If the average effect of 2 and 4 mg of brexpiprazole versus placebo was statistically significant (p≤0.05), comparisons for each individual dosage versus placebo were tested. Only if both the 2- and 4-mg dosages were statistically significant in favor of brexpiprazole versus placebo (p≤0.05) for the primary efficacy endpoint measure was the key secondary efficacy measure tested by using the same two-step strategy. Cohen’s d effect size for the primary and key secondary efficacy measures was calculated as the treatment-placebo difference divided by the pooled standard deviation. Statistical analysis of the 0.25-mg brexpiprazole dosage versus placebo was exploratory. Mixed model for repeated measures analysis was applied to changes from baseline in Personal and Social Performance scale score, PANSS subscale scores, PANSS excited component score, PANSS Marder et al. factor scores (
17), body weight, and extrapyramidal symptom scale scores. CGI improvement score at week 6 was analyzed by using the Cochran-Mantel-Haenszel row mean scores test, controlled for site. Responder rates and discontinuation rates due to lack of efficacy were analyzed by the Cochran-Mantel-Haenszel general association test. The number needed to treat for responder rates was calculated as 100/absolute risk reduction. Least squares mean change in body weight at week 6 was derived from an analysis of covariance model with treatment as factor and baseline value as covariate, on observed case data. Mean changes from baseline to the last visit in laboratory measurements were analyzed using analysis of variance with treatment as an effect.
Results
All 636 randomly assigned patients received study medication, and they comprised the safety population (see Figure S2 in the online data supplement). A total of 623 patients were included in the efficacy population, of whom 87, 180, and 178 patients received 0.25, 2, and 4 mg of brexpiprazole respectively, and 178 received placebo.
Demographic and baseline clinical characteristics were similar in the four treatment groups (
Table 1). The patients were markedly ill at entry to the study, with an overall mean PANSS total score of 95.2 and a CGI severity score of 4.9. All patients had previously experienced acute schizophrenia exacerbations requiring treatment.
Almost all patients were taking antipsychotics before the study (579 of 636, 91.0%), while 449 of 636 (70.6%) and 73 of 636 (11.5%) were taking anxiolytics/hypnotics and antidepressants, respectively. During the study, lorazepam was the most frequently used protocol-defined rescue medication, taken by 46 of 90 (51.1%), 94 of 182 (51.6%), and 82 of 180 (45.6%) patients in the 0.25-, 2-, and 4-mg brexpiprazole groups, respectively, and 82 of 184 (44.6%) in the placebo group.
Overall, 410 patients completed the study: 56 of 90 (62.2%), 124 of 182 (68.1%), and 121 of 180 (67.2%) in the 0.25-, 2-, and 4-mg brexpiprazole groups, respectively, compared with 109 of 184 (59.2%) in the placebo group (see Figure S2 in the online data supplement).
Efficacy
The average effect of the 2- and 4-mg brexpiprazole dosages on the primary efficacy endpoint measure was statistically significant compared with placebo (p<0.0001), allowing subsequent comparison of individual dosage groups.
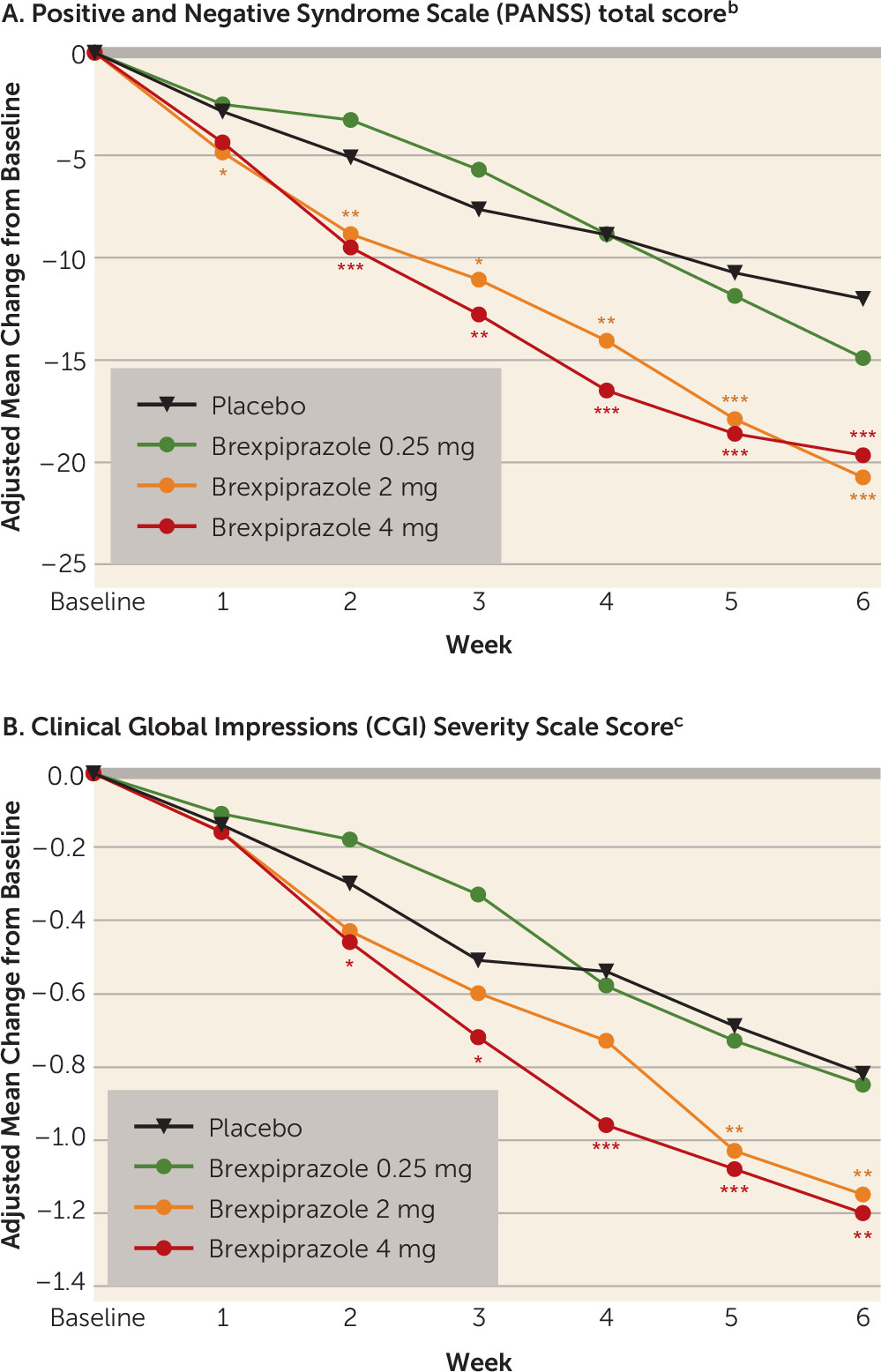
Patients in the 2- and 4-mg brexpiprazole groups had statistically significantly greater mean improvements in PANSS total score than the placebo group at week 6 (
Table 2). At week 6, compared with placebo, the treatment differences were –8.72 (Cohen’s d: 0.41, p<0.0001) and –7.64 (Cohen’s d: 0.36, p=0.0006) for the 2-mg and 4-mg brexpiprazole dosages, respectively. The difference between brexpiprazole and placebo in mean change from baseline reached statistical significance at week 1 in the 2-mg group and at week 2 in the 4-mg group, and the effect was maintained throughout the remainder of the study (
Figure 1A).
The average effect of the 2- and 4-mg brexpiprazole dosages, compared with placebo, on the key secondary efficacy endpoint measure was statistically significant (p=0.0006), allowing subsequent comparison of individual dosage groups.
The mean change from baseline at week 6 in the CGI severity total score was statistically significantly greater in both the 2- and 4-mg brexpiprazole groups than in the placebo group (
Table 2,
Figure 1B). Improvements (p<0.05) from baseline to week 6 that were significantly greater in the 2-mg and 4-mg brexpiprazole groups than in the placebo group were seen in the following secondary efficacy measures: PANSS positive and negative subscale scores, PANSS excited component score, and PANSS Marder et al. factor scores (
17) relating to positive and negative symptoms, disorganized thought, and uncontrolled hostility/excitement (
Table 2). Mean change from baseline at week 6 in the Personal and Social Performance scale score was greater than that for placebo (p<0.05) in the 2-mg brexpiprazole group (
Table 2) but not in the 4-mg group (p=0.06). Improvement at week 6 evaluated by the CGI improvement score was greater in the 2- and 4-mg brexpiprazole groups than in the placebo group (p<0.05,
Table 2). Responder rates were also higher in the 2-mg and 4-mg brexpiprazole groups than in the placebo group (p<0.05), whether response was defined as improvement in PANSS total score of ≥30% (prespecified analysis; number needed to treat: 6 and 7) or as an improvement in score of ≥20%, ≥40%, or≥50% (exploratory analyses; number needed to treat: 6 to 8) (
Table 2). Fewer patients discontinued because of lack of efficacy in the 4-mg brexpiprazole group than in the placebo group (p=0.02) (
Table 2). In the 0.25-mg brexpiprazole group, there were only minimal changes from baseline at week 6 in the primary and secondary efficacy measures (
Table 2).
Safety and Tolerability
The overall incidence of treatment-emergent adverse events was lower in the three brexpiprazole groups (48.9%−56.7%) than in the placebo group (62.0%) (
Table 3). The same was true for discontinuation due to adverse events (8.2%−13.3% versus 17.4%). Most treatment-emergent adverse events with placebo were related to the underlying condition. Akathisia was more frequently reported in the 2- and 4-mg brexpiprazole groups than in the placebo group (4.4% and 7.2% versus 2.2%, respectively) (
Table 3). Akathisia occurred most often during the first 3 weeks of treatment; all incidences were mild or moderate in severity, and none resulted in treatment discontinuation. The incidences of other activating (restlessness, insomnia, anxiety) and sedating (somnolence, fatigue, sedation) treatment-emergent adverse events in patients receiving brexpiprazole were similar to or lower than the rates in patients receiving placebo (
Table 3). The most frequently reported serious adverse events in all treatment groups were psychiatric disorders (schizophrenia, psychotic disorder). There were no deaths during the study.
Increased body weight was reported as a treatment-emergent adverse event by 2.7% and 3.9% of the patients who received 2 and 4 mg of brexpiprazole, respectively, versus 1.6% of the placebo patients. Mean body weight change at week 6 was 1.45 kg and 1.28 kg for the 2- and 4-mg brexpiprazole groups and 0.42 kg for the placebo group; the least squares mean differences from placebo were 1.03 kg for the 2-mg brexpiprazole group (p=0.03) and 0.86 kg for the 4-mg group (p=0.07). An increase in body weight of ≥7% from baseline at any visit was seen in 8.8% and 9.0% of the 2- and 4-mg brexpiprazole groups and 4.4% of the placebo patients.
Changes in fasting-state metabolic measurements are shown in
Table 3. Slight increases occurred in total cholesterol and high-density lipoprotein cholesterol from baseline to the last visit for 2 mg and 4 mg of brexpiprazole, compared with slight decreases in the placebo group, but these changes were neither clinically relevant nor statistically significant. There were slight increases in low-density lipoprotein cholesterol and triglycerides in the 4-mg brexpiprazole group, but these differences were also not clinically relevant. All four groups had minimal increases in glucose. Shifts to abnormal fasting lipid or glucose values were similar in the brexpiprazole and placebo groups, with no significant difference between the groups, as shown in
Table S1 in the online data supplement. Metabolic-related treatment-emergent adverse events were reported by three brexpiprazole patients: diabetes mellitus (0.25 mg), hypertriglyceridemia (4 mg), and increased glycosylated hemoglobin level (4 mg). Treatment-emergent metabolic syndrome (three or more criteria) occurred in none of 144 placebo patients and seven of the brexpiprazole patients: 3.0% (2/67) of those taking 0.25 mg, 1.5% (2/136) of those taking 2 mg, and 2.3% (3/132) of those taking 4 mg.
Mean prolactin concentrations increased from baseline to the last visit in both female and male patients in the 4-mg brexpiprazole group, while reductions were seen in the 2-mg brexpiprazole group and the placebo group (
Table 3). Shifts to abnormal prolactin values were similar in the placebo and brexpiprazole groups, without significant differences between the groups (online Table S1). Hyperprolactinemia was reported as a treatment-emergent adverse event for one patient in the 2-mg brexpiprazole group.
The mean changes in QT interval from baseline to the last visit, as corrected by Bazett’s formula, were 1.1 and 2.3 msec for 2 and 4 mg of brexpiprazole, respectively, compared with 3.1 msec for placebo (
Table 3).
There were no consistent differences between treatment groups in clinical laboratory results (with no significant differences between the groups, online Table S2), vital signs, and ECG measures. Six patients (two each in the 2-mg brexpiprazole group, the 4-mg group, and the placebo group) discontinued the study because of liver-related adverse events (“drug-induced liver injury,” “hepatic enzyme increased,” or “liver function test abnormal”).
Changes from baseline to the last visit in scores on the extrapyramidal symptom scales were minimal in all treatment groups, without statistically significant treatment differences (
Table 4).
The occurrence of suicidal ideation or behavior, as recorded on the Columbia-Suicide Severity Rating Scale, was low. One patient in the 2-mg brexpiprazole group reported suicidal behavior and serious active suicidal ideation at week 1. A different patient in the 2-mg brexpiprazole group had active suicidal ideation at week 1. One placebo patient reported suicidal behavior.
Discussion
In this 6-week randomized, double-blind, placebo-controlled study of markedly ill adults with acutely exacerbated schizophrenia, brexpiprazole at a dosage of either 2 or 4 mg/day resulted in statistically significantly greater improvement than placebo as indicated by the primary outcome measure, change in PANSS total score. Statistically significant improvement was seen in these two brexpiprazole groups within 1–2 weeks of initiating treatment and was maintained throughout the treatment period. The 2- and 4-mg doses were reached by days 5 and 8, respectively. While titration to 4 mg on day 8 could be perceived as relatively late, the difference between the 2-mg arm and placebo reached statistical significance for the primary outcome measure at week 1, with the difference from placebo in the 4-mg arm being close to statistical significance at day 7 (p=0.06), i.e., prior to reaching the 4-mg dose. The key secondary efficacy endpoint measure, mean change from baseline at week 6 in CGI severity total score, was also statistically significantly greater than the change with placebo in the 2- and 4-mg brexpiprazole groups. Other secondary efficacy measures supported the results of the primary and key secondary analyses. Brexpiprazole at dosages of 2 and 4 mg demonstrated efficacy in treating both positive and negative symptoms of schizophrenia, and they reduced agitation as measured by the PANSS excited component. Rates of response (≥30% improvement in PANSS total score or CGI improvement rating of 1 or 2) were higher with the 2- and 4-mg brexpiprazole dosages than with placebo (p=0.0004 and p=0.004, respectively). The 0.25-mg dosage of brexpiprazole did not have any clinically relevant effects on any of the efficacy measures, supporting its purpose in the study, i.e., to explore the lower end of the effective dosage range.
The 2- and 4-mg dosages demonstrated efficacy in four of five PANSS dimensions as defined by Marder et al. (
17): positive symptoms, negative symptoms, disorganized thought, and uncontrolled hostility/excitement. Although brexpiprazole had a limited effect on the anxiety/depression dimension, the enrollment criteria were not designed to select patients with marked levels of anxiety or depression.
The superiority of 2 and 4 mg of brexpiprazole in change in PANSS total score compared with placebo was not only statistically significant but also clinically relevant, indicated by a significantly greater change in CGI severity score as well as Cohen’s d effect sizes of 0.36 to 0.41 for PANSS total score. Moreover, the numbers needed to treat for response (≥30% improvement in PANSS total score or CGI improvement rating of 1 or 2) were 6 for 2 mg and 7 for 4 mg. Both the effect sizes and numbers needed to treat are within the midrange across examined antipsychotics (
4,
24). However, one needs to be careful with indirect comparisons of effect size in placebo-controlled trials across antipsychotics, as an increase in placebo response has been observed (
25). In fact, the 12-point improvement in PANSS total score with placebo in the current study is on the higher side when compared with changes in other regulatory trials of second-generation antipsychotics (
26). Thus, the effect size may be a conservative estimate when compared with those for antipsychotics studied previously, when placebo response was less marked; yet efficacy relative to that of other antipsychotics will need to be tested in future head-to-head trials.
In addition to its demonstrated efficacy, brexpiprazole was well tolerated in this study and had a high completion rate. Notably, more patients randomly assigned to placebo than brexpiprazole discontinued the study because of lack of efficacy or adverse events. Many of the adverse events reported by patients in the placebo group were related to their underlying condition, reported as worsening of schizophrenia. Incidences of activating and sedating adverse events were low and comparable in the brexpiprazole and placebo groups. Consistent with this finding, changes in extrapyramidal symptom scale scores were minimal. A meta-analysis of second-generation antipsychotics has suggested that there are differences between agents in their ability to induce extrapyramidal symptoms (
27), although the only currently available dopamine D
2 partial agonist, aripiprazole, was reported to have a relatively low risk (
4). In this study, the incidence of akathisia was low (4.4% and 7.2% at brexpiprazole doses of 2 and 4 mg, respectively). A moderate increase in body weight in the brexpiprazole groups over 6 weeks was noted, with a difference from placebo of 1 kg or less, but there was no evidence of statistically significant or clinically relevant adverse effects on metabolic measures and prolactin when compared with those for placebo. This finding is important, given that metabolic side effects are a serious concern with some second-generation antipsychotics, namely clozapine, olanzapine, and quetiapine (
5,
28,
29). Preliminary data on newer second-generation antipsychotics suggest that asenapine and iloperidone may also have weight gain potential and metabolic risk, at least in the short term (
30). Some antipsychotics, particularly sertindole and ziprasidone, have been associated with QTc prolongation (
4). However, like lurasidone (
4), brexpiprazole actually resulted in numerically lower QTc changes than placebo when the QTc interval is corrected by using Bazett’s formula. Overall, the tolerability profile of brexpiprazole appears to be consistent with its pharmacological profile, whereby it shows a balanced 5-HT
2A and 5-HT
1A receptor binding affinity relative to D
2, with less intrinsic activity at the D
2 receptor than aripiprazole and moderately low affinity for receptors that have been associated with sedation and weight gain (
8).
Brexpiprazole is also in clinical development for adjunctive treatment of patients with major depressive disorder and inadequate response to antidepressants. Phase 2 and 3 studies have indicated that adjunctive brexpiprazole is effective and well tolerated in this population (
31,
32). Additional studies are under way for agitation associated with dementia of the Alzheimer's type (
33,
34) and as an adjunct to paroxetine or sertraline in subjects with posttraumatic stress disorder (
35).
The results of this study need to be interpreted within its limitations. These include the lack of an active comparator, short study duration, and inclusion of patients with schizophrenia without other psychiatric comorbidities. However, this study was designed as an acute, short-term regulatory approval study in which comorbidities were excluded to isolate the efficacy signal for schizophrenia symptoms. Clearly, longer-term studies that include a comparator, as well as patients with common comorbid conditions, will be needed to further evaluate the efficacy, safety, and effectiveness of brexpiprazole in the treatment of patients with schizophrenia commonly encountered in clinical care and in comparison with other first-line antipsychotics.
In conclusion, in this 6-week randomized, double-blind, placebo-controlled study, brexpiprazole at doses of 2 mg and 4 mg once daily demonstrated statistically significant efficacy compared with placebo and good tolerability for patients with an acute exacerbation of schizophrenia.
Acknowledgments
The authors thank the investigators at the study sites and the subjects who participated in this study.