The pathophysiology of posttraumatic stress disorder (PTSD) has been heavily examined within the past several decades using different imaging modalities such as positron emission tomography (PET) and functional MRI (fMRI) (
1–
7). In the laboratory, a number of experimental paradigms have been employed to uncover the dysfunctional brain circuits that contribute to persistent fear and anxiety symptoms commonly observed in PTSD patients. Pavlovian fear conditioning and extinction is one of those paradigms that has been studied translationally across various nonhuman species as well as in PTSD patients (
5,
8). The advantages and relevance of this model to PTSD are twofold. First, fear conditioning is relevant to the cause of PTSD. Second, exposure-based therapy, an effective form of PTSD treatment, relies heavily on the principles of fear extinction and its retention (recall). Hence, further examination of the brain network underlying fear extinction could improve our understanding of PTSD and contribute to improved PTSD therapies (
9).
During fear conditioning, a neutral cue predicts an aversive event that causes an unconditioned fear response. After several trials, an association is formed between the neutral cue and the negative outcome, and the presentation of the neutral cue alone comes to elicit conditioned fear responses. Subsequent multiple presentations of the cue without any reinforcement lead to fear extinction (
10). Animal and human neuroimaging studies of fear extinction learning and its recall have identified an essential network that includes the amygdala, the hippocampus, the ventromedial prefrontal cortex (vmPFC), and the dorsal anterior cingulate cortex (dACC) (
11–
17). Compared with trauma-exposed individuals, patients with PTSD show normal within-session extinction learning but impaired extinction recall when tested after a delay (
18–
20), as well as an impaired ability to inhibit fear in the presence of a safety signal (
21). The impairments in extinction recall and contextual safety signal processing observed in PTSD have been associated with vmPFC and hippocampus hypoactivation and with dACC and amygdala hyperactivation (
20,
22–
24). Dysfunctional resting-state activity in these same brain regions has also been reported. Specifically, studies examining resting regional cerebral blood flow in individuals suffering from PTSD have reported hyperactive amygdala (
25,
26) and dACC (
27) but hypoactive medial prefrontal cortex (
26).
Although much has been learned about neural circuits contributing to the pathophysiology of PTSD, knowledge gaps remain. The relationships among PTSD symptoms, resting-state alterations, and abnormal functional brain activity have not been examined within the same individuals. It would be informative to learn whether alterations in resting regional brain functioning are related to variations in extinction recall in PTSD. No previous studies have measured both resting brain glucose metabolism and fear extinction-related functional activation within the same PTSD subjects. In addition, few previous studies have examined the effect of trauma exposure per se on resting activity and activation in the extinction network. The majority of published studies have employed either trauma-exposed non-PTSD groups or non trauma-exposed groups, but not both groups.
In the present study, trauma-exposed individuals with PTSD and trauma-exposed non-PTSD individuals underwent a clinical interview to determine diagnostic status, symptom severity, and level of functioning, as measured by the Global Assessment of Functioning Scale (GAF). Using
18F-fluorodeoxyglucose (FDG) PET, resting brain glucose metabolism was assessed in all participants. Approximately 4 days later, participants underwent a 2-day fear conditioning and extinction paradigm during fMRI. Skin conductance responses were measured as a psychophysiological index of fear, and fMRI blood-oxygen-level-dependent (BOLD) signal was measured as a marker of functional regional brain activation. Previously published data on trauma-unexposed healthy individuals who had undergone the same paradigm were also used for between-group comparisons (
28). We hypothesized that in the trauma-exposed groups, resting glucose metabolism in the nodes of the fear extinction network would be related to clinical outcomes and to functional activations in the same brain regions during extinction recall. We also hypothesized the presence of differences among the three groups in resting glucose metabolism and extinction recall functional activations in the amygdala, hippocampus, dACC, and vmPFC.
Method
Participants
Forty-four right-handed individuals aged 18 to 65 were recruited from the community. Of these, 24 met criteria for a current PTSD diagnosis and 20 were trauma-exposed but had no PTSD (the TENP group). For the PET and fMRI between-group analyses, we also included 21 healthy comparison subjects who had not been exposed to trauma, for whom the PET-fMRI results have already been published (
28). For a full description of the sample and a list of exclusion criteria, see the Supplemental Methods section and Table S1 in the
data supplement that accompanies the online edition of this article.
Procedure
All subjects provided written informed consent for participation after receiving a full explanation of the procedures, a detailed description of which is provided in Supplemental Methods in the
online data supplement. Briefly, participants were first interviewed by the study diagnostician. They then underwent a PET-FDG scan during which brain glucose metabolism was assessed as a measure of resting brain activity (
28). Approximately 4 days later, they took part in a 2-day fear conditioning and extinction paradigm during fMRI (
11,
17,
18,
23,
24,
28). This paradigm consisted of a habituation phase where both contexts A and B were presented, followed by a conditioning phase in context A, during which the participant viewed one of three different-colored lamps (CS+s), two of which were partially reinforced by an electrical stimulation to the index finger (US). The third colored lamp (CS−) was never reinforced. Immediately after conditioning, extinction training took place in context B. One of the CS+ stimuli (designated the CS+E) was repeatedly presented (along with the CS−) in the absence of the US. The following day, extinction recall (retention) was tested in context B by presenting the CS+E, the nonextinguished CS+ (designated the CS+NE), and the CS−.
Data Processing
Imaging.
All coordinates reported are based on the Montreal Neurological Institute (MNI) system. Region-of-interest analyses were performed for both PET and fMRI. The Anatomical Automatic Labeling atlas (
29) was used to define the amygdala and the hippocampus, and data were extracted bilaterally. Based on our previous studies (
17,
28,
30), the vmPFC was defined as a 5-mm sphere centered at MNI x, y, z coordinates 5, 35, −13, and the dACC was defined as a 5-mm sphere centered at coordinates 2, 22, 29. For details on the treatment and processing of the PET-FDG and fMRI data, see Supplemental Methods in the
data supplement.
Psychophysiological data.
Skin conductance response to each stimulus was computed by subtracting the mean skin conductance level observed during the last 2 seconds of context alone preceding CS onset from the maximal skin conductance level reached during CS presentation. All skin conductance response values were square-root-transformed prior to analysis. To evaluate extinction recall, an extinction retention index was computed for each individual, using the following formula: (100 − [mean skin conductance response to the first four CS+E trials during recall / maximum skin conductance response reached during conditioning for this same cue]) × 100.
Clinical data.
PTSD symptom severity was calculated by adding the frequency and intensity scores for each item on the Clinician-Administered PTSD Scale (CAPS) for DSM-IV. A total CAPS severity score and scores for each cluster of symptoms (re-experiencing, avoidance/numbing, and hyperarousal) were calculated. The GAF was administered in both trauma-exposed groups. Because the CAPS scores were all close to zero in the TENP group, the GAF offered the advantage of measuring functioning in trauma-exposed participants. Therefore, GAF scores were analyzed for both trauma-exposed groups, whereas CAPS scores were analyzed only in the PTSD group.
Data Analysis
In order to determine whether glucose resting metabolism within the a priori defined nodes of the fear extinction network predicts functional activation in that circuit during extinction recall, FDG values within the predefined regions of interest were used as regressors for the fMRI contrast (whole-brain voxel-wise analysis). This was done for each of the trauma-exposed groups separately. Because the same analytic approach has already been published for the healthy comparison group, we did not include this group in these analyses. For the early CS+E versus early CS+NE BOLD contrast, the following extracted PET FDG values were used as a single regressor in separate analyses: 1) average of left and right amygdala, 2) average of left and right hippocampus, 3) dACC, and 4) vmPFC. An initial criterion of p<0.005, with a minimum of 10 contiguous voxels, was used to detect positive or negative correlations between resting FDG measures and BOLD signal during extinction recall in four brain regions: the amygdala, the hippocampus, the dACC, and the vmPFC. Clusters detected within these regions that survived small-volume family-wise error correction (p<0.05) were considered significant. After conducting the PET-fMRI analyses within each group, follow-up interaction tests were conducted to test between-group differences in this measure (PET-fMRI correlations). A threshold of p<0.005 with 10 contiguous voxels was applied.
In order to investigate differences between the TENP and healthy comparison groups, for the four fear extinction network regions of interest (averaged left and right amygdala, averaged left and right hippocampus, dACC, and vmPFC, separately), analyses of covariance (ANCOVAs) were performed on the extracted PET resting glucose metabolism data, using group as the between-subject factor and age and years of education as covariates (given that these variables show significant group differences—see the data supplement). If the ANCOVA revealed a significant main effect of group at a p<0.05 level, Tukey least significant difference post hoc tests were run on the estimated marginal means (controlled for age and years of education) to identify significant group differences.
Results
Sample Characteristics
The sample characteristics are summarized in Table S1 in the online data supplement. A Fisher exact probability test confirmed that sex did not significantly differ among the three groups. One-way analyses of variance revealed the presence of a significant group difference for age (F=8.0, df=2, 61, p=0.001) and years of education (F=13.1, df=2, 57, p<0.001). Scheffé post hoc t tests confirmed that on average the healthy comparison group had significantly more years of education than the two trauma-exposed groups and was younger than the PTSD group (p values ≤0.05). All analyses investigating differences among these three groups therefore included age and years of education as covariates. An independent-samples t test was performed on CAPS scores between the two trauma-exposed groups and confirmed significantly higher PTSD symptom severity in the PTSD group (p<0.001).
Psychophysiological Data
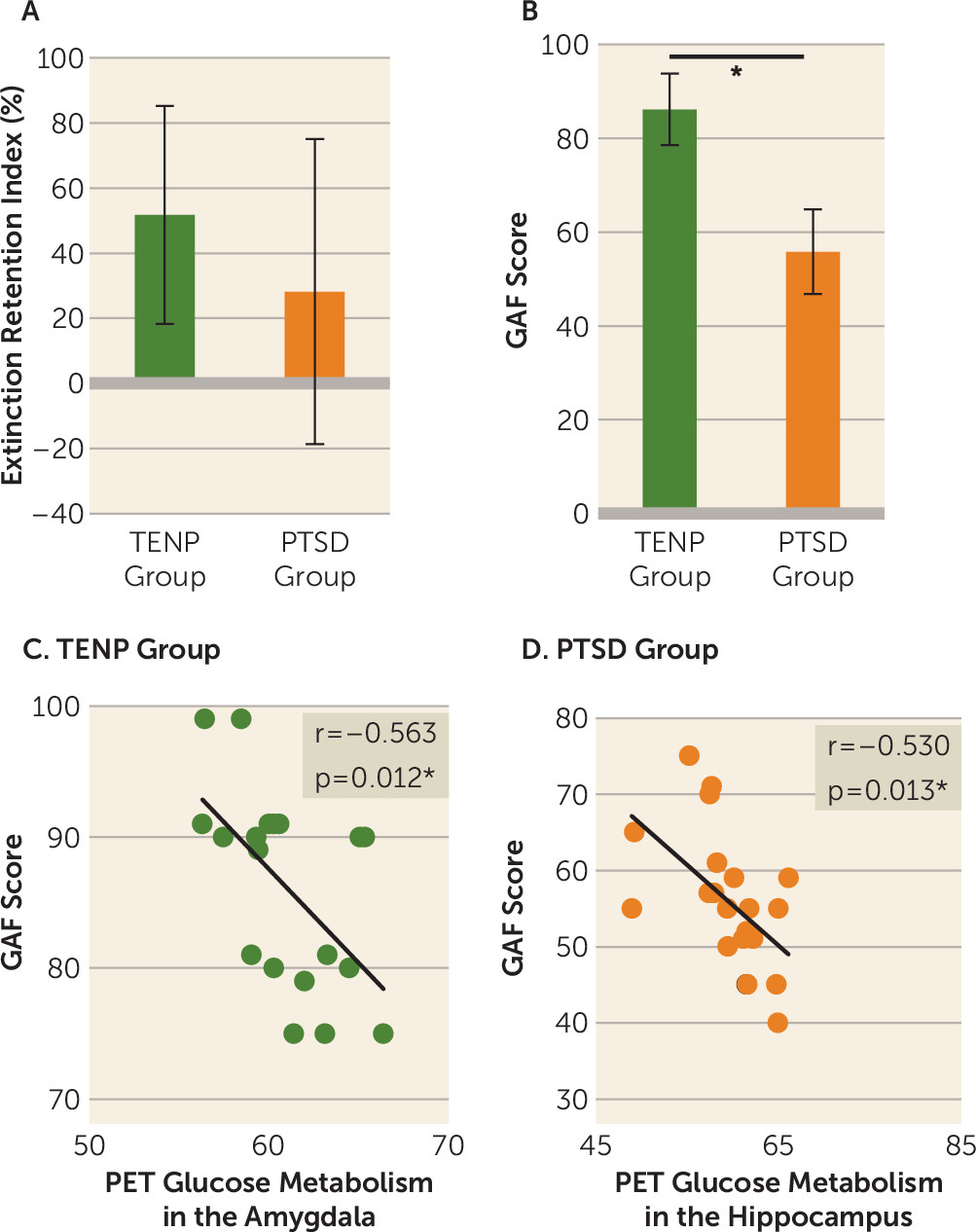
An independent-samples t test performed on extinction retention index revealed that the PTSD group tended to exhibit a lower extinction retention index than the TENP group (t=1.79, df=26, p=0.086) (
Figure 1A). After applying a Bonferroni correction for multiple comparisons (0.05/4 regions of interest=0.0125), no correlations between extinction retention index and PET data from each region of interest survived in either trauma-exposed group.
Clinical Data
An independent-samples t test confirmed significantly lower GAF scores in the PTSD relative to the TENP group (t=11.40, df=38, p<0.001) (
Figure 1B). We correlated the GAF scores with resting metabolism from each region of interest for both trauma-exposed groups separately, using a Bonferroni correction (0.05/4 regions of interest=0.0125). PET resting metabolism in the amygdala was negatively correlated with GAF scores in the TENP group (r=−0.563, df=17, p=0.012) (
Figure 1C), and PET resting metabolism in the hippocampus was negatively correlated with GAF scores in the PTSD group (r=−0.530, df=19, p=0.013) (
Figure 1D).
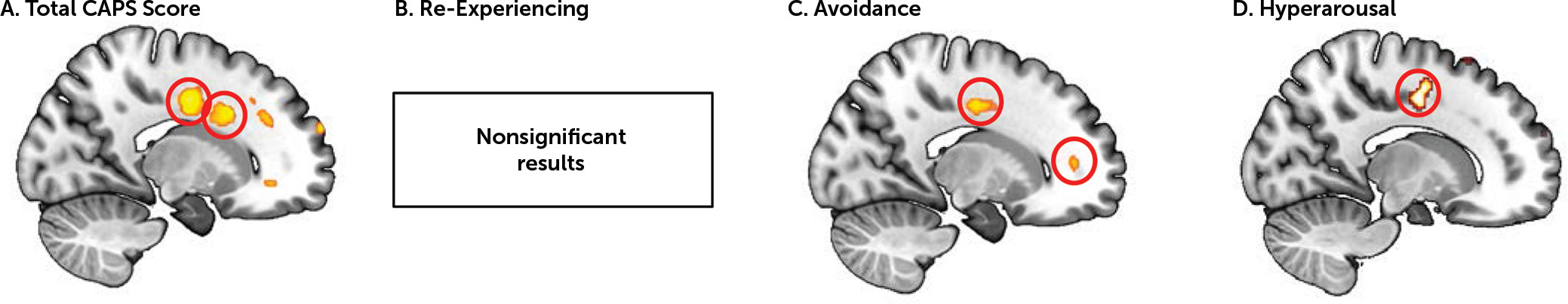
When correlating PTSD symptom severity with resting metabolism extracted from the four regions of interest of the fear extinction network in the PTSD group separately, no significant results were found. We therefore performed a supplemental whole-brain analysis to assess the relationship between glucose metabolism in the nodes of the fear extinction network and PTSD symptoms. Whole-brain analysis performed on PET data using total CAPS score as a regressor revealed a positive association in two different areas of the dACC (x, y, z=18, 14, 32; cluster size=410; t=4.05, df=17, family-wise-error-corrected p [p
FWE]=0.016; and x, y, z=18, −8, 38; cluster size=410; t=4.22, df=17, p
FWE=0.012) (
Figure 2A). No associations were found with re-experiencing symptoms (
Figure 2B). Avoidance symptoms were positively associated with resting metabolism in the dACC (x, y, z=14, −6, 42; cluster size=62; t=3.72, df=17, p
FWE=0.007) and the rostral ACC (rACC) (x, y, z=16, 44, 8; cluster size=32; t=3.28, df=17, p
FWE=0.016) (
Figure 2C). Finally, hyperarousal scores were positively associated with dACC resting metabolism (x, y, z=16, −6, 44; cluster size=70; t=3.44, df=17, p
FWE=0.014) (
Figure 2D).
PET Predicting BOLD During Extinction Recall in Trauma-Exposed Groups
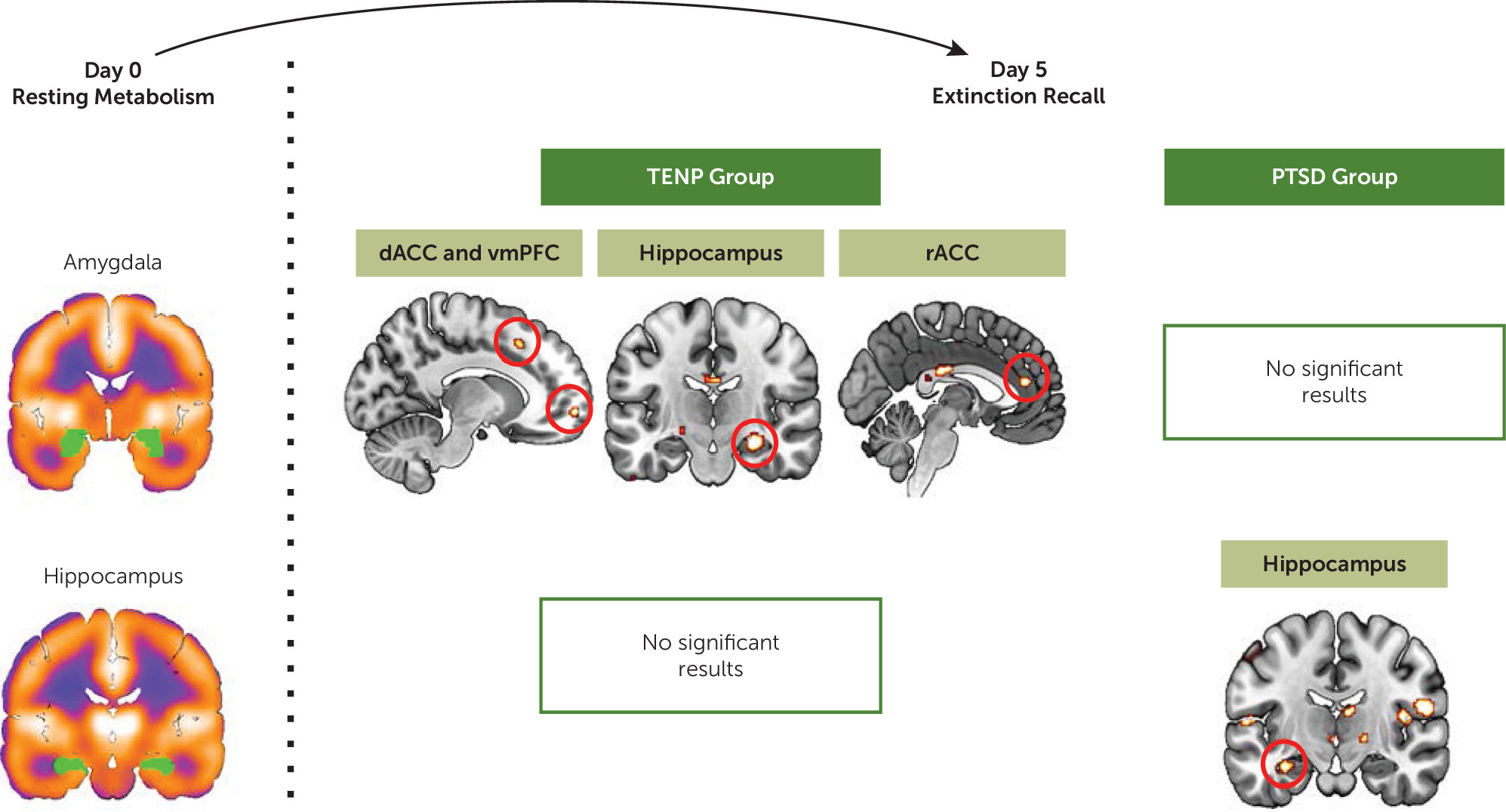
Amygdala.
For the TENP group, resting metabolism in the amygdala positively predicted activations in the following regions (
Figure 3): dACC (x, y, z=−12, 20, 46; cluster size=10; t=3.48, df=13, p
FWE=0.007), vmPFC (x, y, z=−12, 60, −6; cluster size=19; t=4.10, df=13, p
FWE=0.012), hippocampus (x, y, z=28, −18, −16; cluster size=79; t=5.56, df=13, p
FWE=0.001), and rACC (x, y, z=−2, 40, 16; cluster size=27; t=4.06, df=13, p
FWE=0.005). No significant results were found in the PTSD group.
Hippocampus.
For the TENP group, no significant results were found (
Figure 3). For the PTSD group, hippocampus resting glucose metabolism predicted its own activation (x, y, z=−32, −14, −20; cluster size=27; t=3.67, df=16, p
FWE=0.008).
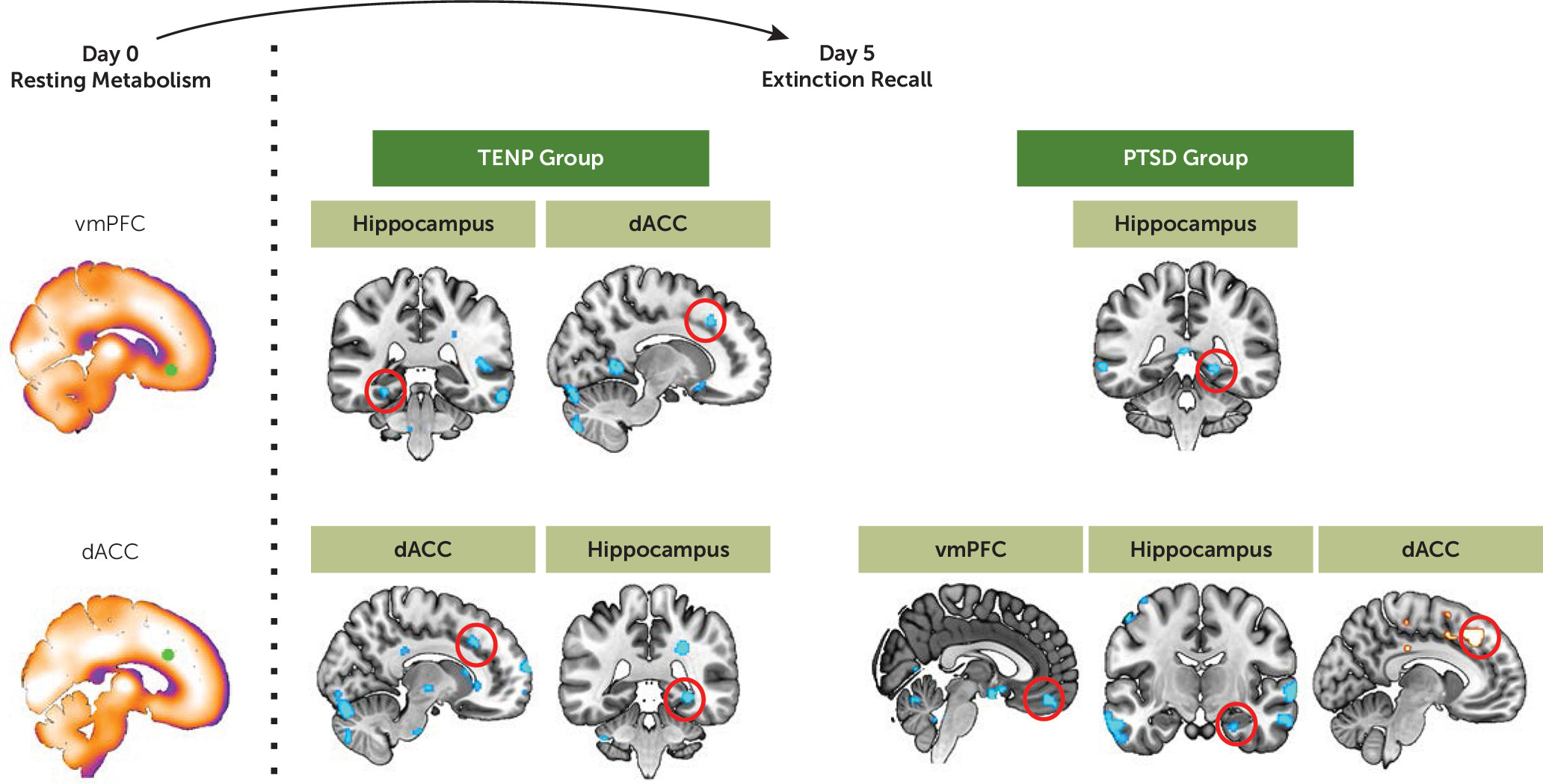
vmPFC.
For the TENP group, vmPFC resting metabolism negatively predicted activation in the posterior hippocampus (x, y, z=−28, −36, −12; cluster size=11; t=3.85, df=13, p
FWE=0.005) and in the dACC (x, y, z=12, 24, 38; cluster size=19; t=3.62, df=13, p
FWE=0.009) (
Figure 4). For the PTSD group, vmPFC resting metabolism negatively predicted posterior hippocampus activations (x, y, z=22, −42, −4; cluster size=19; t=4.02, df=16, p
FWE=0.003).
dACC.
For the TENP group, dACC resting metabolism negatively predicted its own activation (x, y, z=12, 26, 36; cluster size=26; t=3.54, df=13, p
FWE=0.013) and activation in the posterior hippocampus (x, y, z=30, −38, −8; cluster size=34; t=3.62, df=13, p
FWE=0.013) (
Figure 4). For the PTSD group, dACC resting metabolism positively predicted its own activation (x, y, z=−8, 10, 42; cluster size=24; t=3.28, df=16, p
FWE=0.015) and negatively predicted activation in the vmPFC (x, y, z=14, 38, −30; cluster size=372; t=5.67, df=16, p
FWE=0.002) and the hippocampus (x, y, z=24, −10, −26; cluster size=19; t=3.29, df=16, p
FWE=0.012).
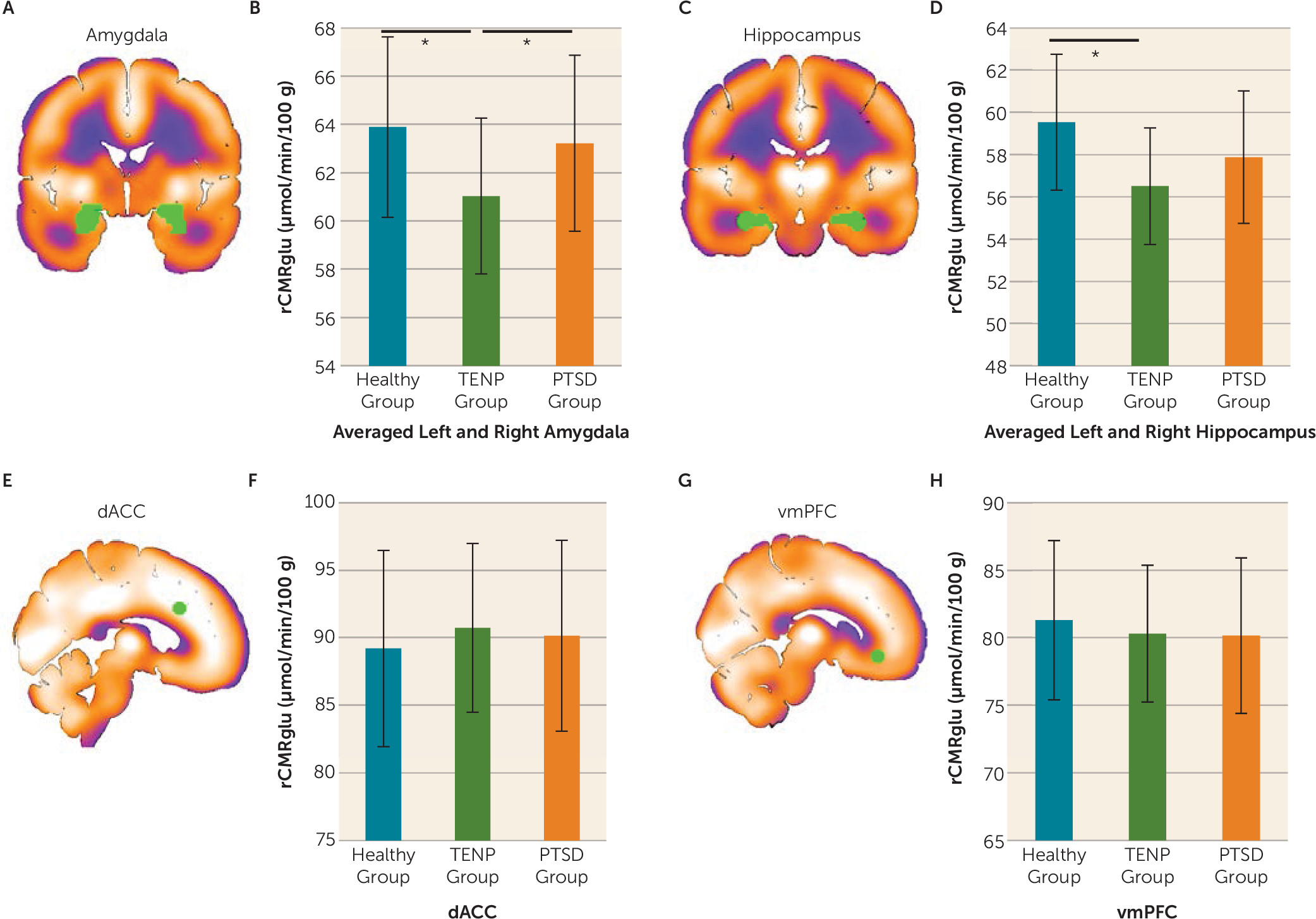
Between-Group PET Differences
We then added the previously published data from the healthy comparison subjects (
Figure 5). One-way ANCOVAs performed on FDG values from the four nodes of the fear extinction network yielded a main effect of group for the amygdala bilaterally (F=3.97, df=2, 55, p=0.025) and the hippocampus bilaterally (F=3.26, df=2, 55, p=0.012). Post hoc tests revealed that the TENP group had lower resting metabolism in the amygdala relative to the PTSD and healthy comparison groups (p values ≤0.044) as well as lower hippocampal resting metabolism relative to the healthy comparison group (p=0.004). There was no main effect of group for the dACC and the vmPFC.
For a report of between-group fMRI differences during extinction recall, see the Supplemental Results section of the online data supplement.
Discussion
In a multimodal imaging study, we gathered PET-FDG, fMRI BOLD, psychophysiological, and clinical measures from trauma-exposed individuals with and without a PTSD diagnosis. We also added previously published data from a group of healthy comparison subjects and tested between-group differences within each imaging modality to unveil the effects of trauma exposure and diagnosis on resting metabolism and reactivity of the fear extinction network during extinction recall. When we focused on the trauma-exposed groups, resting metabolism in the amygdala and the hippocampus was associated with global functioning. In the PTSD group, dACC resting metabolism was associated with PTSD symptom severity and was predictive of activation patterns during extinction recall that have been shown to be characteristic of PTSD psychopathology. At rest and during extinction recall, between-group differences were observed. Lower resting metabolism in the amygdala and the hippocampus was observed in the TENP group, whereas hypoactivation of the vmPFC during extinction recall was observed in the PTSD group. Together, these results suggest that resting metabolism in the nodes of the fear extinction network is correlated with both self-reported and biologically based clinically relevant measures in trauma-exposed individuals. Importantly, the alterations observed in the resting metabolism of the amygdala and hippocampus indicate that trauma-exposed individuals with and without PTSD show long-lasting signs of perturbations in brain function, even in the absence of stimuli probing the fear circuitry or the recall of the trauma.
The inclusion of a non-trauma-exposed healthy comparison group enabled us to examine the effects of trauma exposure and diagnostic status on both resting metabolism and functional activation during extinction recall. Our results showed that the TENP group had lower amygdala resting metabolism when compared with the healthy comparison and PTSD groups and lower hippocampal resting metabolism relative to the healthy comparison group. Notably, PTSD patients have often been compared only to a trauma-exposed non-PTSD group. Therefore, the elevated amygdala resting metabolism is often thought to be an acquired sign of PTSD or to be a pre-existing vulnerability to PTSD. However, in the present study, the resting metabolism of the amygdala and the hippocampus in the PTSD group was similar to that of the healthy comparison group. This suggests that reduced glucose metabolism in the TENP group could reflect a resilience factor (e.g., trauma exposure may lead to reduced resting metabolism in the amygdala and hippocampus only in resilient individuals). Interestingly, in the TENP group, amygdala resting glucose metabolism was negatively associated with level of functioning as measured by GAF scores. This further supports the point that suppressing resting metabolism in the amygdala after trauma exposure may be a key resilience factor to prevent the development of PTSD and to improve functioning. In PTSD, lower hippocampus resting metabolism was associated with better functioning. When we focused on between-group differences during extinction recall, the PTSD group exhibited lower vmPFC activation relative to the TENP and healthy comparison groups. For this comparison, the TENP group did not differ significantly from the healthy comparison group. These data suggest that lower vmPFC activation during extinction recall may be a characteristic of PTSD, rather than of trauma exposure per se. While the comparison between the PTSD and TENP groups replicates previous findings from our group and others (
20,
23,
24), the addition of the healthy comparison group adds an extra layer of information suggesting that trauma exposure per se does not significantly affect the ability to activate the vmPFC during extinction memory recall. Taken together, these results suggest that lower resting metabolism in the subcortical regions seems to be associated with resilience following trauma, and that hypoactivation of the prefrontal cortex during extinction recall is a specific characteristic of the PTSD group.
Previous studies from our group and others have shown that individuals with PTSD exhibit poorer extinction memory recall (
20,
23). This psychophysiological deficit has also been associated with a distinct pattern of brain activation in which the vmPFC and the hippocampus are hypoactive but the dACC is hyperactive. As mentioned earlier, we replicated the finding pertaining to the hypoactivation of the vmPFC during extinction recall. Moreover, we replicated the psychophysiological finding at a trend level by demonstrating lower extinction retention in the PTSD group relative to the TENP group. A key finding of the present study is that for the PTSD group, dACC resting metabolism predicted functional activations during extinction recall that were lower in the vmPFC and hippocampus but higher in the dACC. This suggests that dACC resting metabolism could potentially serve as a predictor of the typical neural activation patterns observed in individuals with PTSD during extinction recall. Notably, in whole-brain analyses with PTSD symptom severity in the PTSD group, we found that dACC resting metabolism was positively associated with overall symptom severity, avoidance, and hyperarousal symptoms. Taken together, these data suggest that higher resting metabolism in the dACC is associated with the altered pattern of activation during extinction recall that has been shown to be characteristic of PTSD and is also associated with greater PTSD symptom severity. After conducting interaction analyses to compare differences in correlation patterns between groups (see Table S2 in the
online data supplement), we found that dACC resting metabolism was the node that yielded most of the interactions, suggesting that resting metabolism in that brain region predicts more distinct patterns between the two trauma-exposed groups.
Twin studies (
27,
31) have suggested that resting glucose metabolism and functional activation within the dACC are elevated in PTSD and may reflect a familial vulnerability factor for developing the disorder rather than an acquired feature of trauma exposure or PTSD. Our data are in apparent contradiction with those findings, given that we did not observe a significant between-group difference in dACC resting metabolism. One possible reason is that our sample was relatively young (mean ages of 39.8 years and 34.1 years for the PTSD group and the TENP group, respectively) and consisted of men and women exposed to different types of trauma. In contrast, one of the twin studies (
27) consisted of older men (mean ages of 57.8 years and 57.1 years for the PTSD group and the TENP group, respectively) who were exposed specifically to combat-related trauma. Thus, age at testing, type of trauma, age at trauma onset, and sex differences may all be factors contributing to the apparent discrepancy between findings. In addition, the coordinates used for the dACC in our study are different from those reported in the twin studies. Despite these differences, the data are converging to show that dACC resting metabolism is an important correlate of neural activations and PTSD symptom severity. This supports previous data from one of the twin studies (
27) that not only demonstrated a positive correlation between dACC resting metabolism and symptom severity in individuals with PTSD, but also revealed a positive correlation between dACC resting metabolism in unexposed co-twins and their brothers’ PTSD symptom severity.
The present study’s limitations should be considered when interpreting the data. First, the healthy comparison group was significantly younger than the PTSD group and better educated relative to both trauma-exposed groups. However, controlling for these factors by covariance analysis should have mitigated their influence on our findings. Second, although each group is large enough to allow group comparisons, the sample size does not allow us to divide participants as a function of sex, age, and other demographic variables that may affect the results. This is worthy of note here, given that there are differences between the types of trauma in the TENP and PTSD groups. Larger groups would allow us to disentangle the contributions of such variables. Finally, the sample size for each group was not constant across the various analyses, because of movement in the scanner, data quality, and dropout. These limitations highlight the need for replication of the findings before definitive conclusions can be drawn.
The intricacy of our results reflects the complex influence that trauma exposure and PTSD have on the neural circuitry mediating extinction recall. By incorporating two comparison groups differing in trauma history, our data enabled us to identify distinct patterns of resting metabolism and neural activation as a function of trauma exposure and diagnosis. This allows a better understanding of how trauma exposure affects the brain independently of PTSD symptoms and therefore helps shed light on the biological underpinnings of the disorder. Our findings support the importance of examining resting metabolism in the fear extinction network. In fact, our results highlight the fact that the neural effects of trauma exposure and PTSD diagnosis can be observed at rest, without probing the network with fear-related stimuli. Our findings also highlight the utility of examining resting metabolism in the nodes of the fear extinction network to predict activations in the network during extinction recall. Given the established relationship between extinction and exposure-based therapy and the fact that individuals with PTSD show impairments in extinction recall at the psychophysiological and neural levels, it is crucial to identify predictors of such dysfunctional patterns to better inform treatment. Our data suggest a relationship between dACC resting metabolism and both PTSD symptom severity and the dysregulated pattern of brain activation observed in PTSD during extinction recall. Therefore, we suggest that dACC resting metabolism should be further examined as a potential biomarker for PTSD. Therapeutic interventions or brain stimulation techniques could be studied as potential modifiers of resting brain metabolism. If successful, these targeted techniques could potentially be used to improve clinical outcomes in trauma-exposed individuals.
Acknowledgments
The authors thank Dr. Gregory Quirk, Dr. Roger Pitman, and the members of the Massachusetts General Hospital Behavioral Neuroscience Program for their helpful comments on the manuscript.