Schizophrenia is characterized by positive and negative symptoms and often by cognitive impairment. The overwhelming majority of patients with schizophrenia suffer to some extent from negative symptoms (
1–
3), which persist for most of their lives (
4). Currently available antipsychotic drugs, all of which to varying degrees have antidopaminergic activity, ameliorate positive symptoms in about two-thirds of acutely ill patients. After amelioration or remission of symptoms, most patients are maintained on antipsychotics to reduce risk of relapse.
Antipsychotic drugs such as amisulpride (
5,
6) and, more recently, asenapine (
7) and cariprazine (
8) have been suggested to be beneficial for negative symptoms, but their specificity and advantages in this regard remain debatable (
9,
10). On the other hand, some dopamine D
2 receptor blocking antipsychotic drugs produce secondary negative symptoms, which are not always easy to distinguish from primary negative symptoms (
11). Therefore, according to the Patient Outcomes Research Team schizophrenia guidelines, no pharmacologic treatment for negative symptoms has proved to have sufficient evidence to support a treatment recommendation (
12). Another limitation of the currently available pharmacological treatments is their high incidence of adverse effects such as anhedonia, which often produce and enhance negative symptoms. Depending on the specific agent, these drugs also may produce extrapyramidal symptoms, sedation, increased prolactin secretion, weight gain, and other metabolic abnormalities.
Basic research indicates that pharmacologically manipulated
N-methyl-
d-aspartate (NMDA)/glutamate neurotransmission, via glycine reuptake, may ameliorate negative symptoms (
13). This hypothesis was supported in a clinical trial with bitopertin (
14), but the results were not replicated in a subsequent large multicenter trial (
15). Likewise, apparent efficacy for cognitive impairment and negative symptoms reported in preliminary trials with the nicotinic agonist encenicline (
16) has not been confirmed in larger, well-controlled pivotal trials (
17). Hence, the search for drugs with broader efficacy beyond positive symptoms to include negative symptoms and cognitive functioning, coupled with an acceptable tolerability profile, remains a priority (
18).
MIN-101 is a novel cyclic amide derivative that has high equipotent affinities for sigma-2 and 5-hydroxytryptamine 2A (5-HT
2A) receptors (inhibitory constants [Ki] of 7.53 nmol/L and 8.19 nmol/L for 5-HT
2A and sigma-2, respectively). MIN-101 also shows binding affinity for α
1-adrenergic receptors but low or no affinity for muscarinic, cholinergic, and histaminergic receptors (data on file). Although MIN-101 has no affinities for pre- or postsynaptic dopaminergic receptors, it is probable that sigma-2 receptors are implicated in the modulation of dopamine (
19,
20) and glutamatergic pathways (
21), as well as in calcium neuronal modulation (
22). Recent research on sigma receptors also shows new binding sites, such as the progesterone receptor membrane component 1 (PGRMC1), but these data are still under debate (
23). Taken together, the data suggest that it could be hypothesized that sigma-2 receptors are involved in counteracting dysregulations in key dopamine and glutamate neurotransmitter pathways. It should be noted that some antipsychotic drugs, such as haloperidol, possess sigma-1 binding activities (
23), but the sigma receptors’ role in affecting schizophrenia symptoms has not been elucidated. In an open-label trial with 28 patients suffering from schizophrenia treated with a compound that selectively binds to sigma-1 receptors, the results were uninterpretable and the development was abandoned (
24).
In rodents, MIN-101 has been found to improve impairment of social interaction induced by phencyclidine and impairment of spontaneous alternation behavior induced by MK-801 (data on file). These experiments are hypothesized to be predictive of a therapeutic effect on negative symptoms. Also in rodents, higher doses of MIN-101 have been found to reduce hyperlocomotion induced by apomorphine and by methamphetamine. These models are predictive of antipsychotic effects.
Finally, MIN-101 has demonstrated effects on the conditioned avoidance response test, without reducing the percentage of escapes, consistent with the finding that MIN-101 does not have a sedative effect. In addition, basic science work has indicated that 5-HT
2A receptors may be implicated in schizophrenia (
25); hence, blocking these receptors may contribute to MIN-101’s therapeutic effect.
A phase 2a randomized, placebo-controlled proof-of-concept and hypothesis-generating study was previously conducted with MIN-101 in patients with acute schizophrenia (a score ≥60 on the Positive and Negative Syndrome Scale [PANSS] and a rating ≥4 on the severity item of the Clinical Global Impressions Scale [CGI]). Patients were discontinued from all psychotropic medications and then randomly assigned to receive placebo or MIN-101. Statistically significant improvements in negative symptoms measured by the pentagonal structure model of the PANSS were observed after 12 weeks of treatment (
26).
The phase 2b trial described here was designed to confirm and extend the findings of the phase 2a trial in symptomatically stable schizophrenia patients. All patients provided written informed consent, and the study was approved by the relevant ethics committees and regulatory authorities at each site and was conducted according to Good Clinical Practice guidelines.
Method
Patient Population
The trial enrolled 244 patients 18–60 years of age at 36 sites in six European countries between May 2015 and December 2015. To be eligible, patients had to meet DSM-5 criteria for schizophrenia, confirmed by the MINI International Neuropsychiatric Interview (
27). Patients had to be symptomatically stable according to their treating psychiatrist, and they had to have had negative symptoms for >3 months prior to entering the trial. At baseline, patients had to have at least moderately severe negative symptoms, with a score ≥20 on the “classic” seven-item negative symptom scale of the PANSS (items N1–N7) and, in order to reduce potential dropout, scores <4 on the following PANSS items: excitement, hyperactivity, hostility, suspiciousness, uncooperativeness, and poor impulse control. Patients were excluded if they had a personal or family history of long QT syndrome, if their QTc (Fridericia-corrected) was >430 msec for males and >450 msec for females, or if genotyping indicated that they were poor or intermediate metabolizers for P450 CYP2D6. Patients with a diagnosis of another mental disorder, a significant risk of suicide, an unstable medical disorder, or a history of substance abuse within 3 months of the screening visit or a positive urine test for illicit drugs were also excluded.
Study Design
Eligible patients were withdrawn from depot antipsychotics for at least 1 month. They were then hospitalized and withdrawn from all psychotropic drugs for at least 5 days before randomization. Patients were randomly assigned to receive placebo or oral MIN-101 at 32 mg/day or 64 mg/day, in a 1:1:1 ratio, for 12 weeks. The randomization was conducted centrally by an independent vendor, and all investigators, patients, sponsor staff, and study supervising staff were blind to the assignment at all times during the study. After randomization, patients had to be hospitalized for at least 36 hours and then could remain hospitalized or be discharged at the discretion of the investigator.
No other psychotropic medications were allowed during the 12-week trial except for rescue medications given for insomnia or agitation in doses allowed by local regulations (oral lorazepam, oral zolpidem, or injectable sodium amytal). Anticholinergic medications were discontinued at baseline in all patients but were allowed during the trial to treat emergent extrapyramidal symptoms. Patients who completed the 12-week trial could continue to receive the same dosage of MIN-101 or be switched from placebo to MIN-101 at 32 mg/day or 64 mg/day for 24 additional weeks in an open-label study (data not presented here).
Assessments for efficacy and safety were conducted at baseline before the first dose of medication and at weeks 2, 4, 8, and 12 or on premature termination. In addition, outpatients were contacted by telephone at weeks 6 and 10 to ascertain safety and adherence to study medication.
Outcome Measures
The primary outcome measure was the negative factor score of the PANSS from the pentagonal structure model (items N1–N4, G5–G8, G13, G14) (
28). Secondary outcome measures were PANSS total score, PANSS positive, negative, and general psychopathology scale scores, the individual pentagonal factors of the PANSS, Brief Negative Symptom Scale score (
29), CGI severity and improvement scores, Brief Assessment of Cognition in Schizophrenia (BACS) score (
30), and Calgary Depression Scale for Schizophrenia (CDSS) score (
31). The primary and secondary efficacy outcome measurements reported here were all defined a priori. Safety and tolerability were evaluated by monitoring the frequency, severity, and timing of adverse events, clinical laboratory test results, triplicate 12-lead ECG, vital sign measurements, body weight, the Sheehan–Suicidality Tracking Scale (
32), and the Abnormal Involuntary Movement Scale (AIMS).
Sample Size and Statistical Analysis
The study was powered at 90% with a two-sided significance level of 0.05. Based on the results of the previous study, a robust treatment effect was assumed of an effect size (Cohen’s d) of 0.60 (3 points, with a standard deviation of 5) in the mean change from baseline to week 12 in the PANSS five-factor (pentagonal structure model) negative factor score between either dose of MIN-101 and placebo. Intent-to-treat and per-protocol analyses were performed. Given space constraints, the per-protocol results are presented only for the primary study measure, the PANSS negative factor, and complete reporting is provided for the intent-to-treat analysis.
The primary endpoint analysis, change from baseline to week 12 in PANSS negative factor (pentagonal structure model), was performed using a mixed-effects model for repeated measures with treatment arm, pooled study center visit (by country), and treatment arm-by-visit interaction as fixed effects, patient nested in treatment as a random effect, and baseline value as a covariate. An unstructured covariance matrix was used to model the covariance of within-patient scores. The Kenward-Roger approximation (
33) was used to estimate denominator degrees of freedom. This analysis was performed based on all postbaseline scores using all observed data without imputation of missing values.
Pairwise comparison between the high and low dosages of MIN-101 and placebo was performed. In order to maintain the type I error rate due to multiple comparisons for the primary endpoint at or below 0.05%, the Hochberg procedure was used. This procedure allows testing of the null hypothesis of no treatment difference for both the 64 mg/day and 32 mg/day dosages compared with placebo to be rejected if the largest p value comparing either of the two dosages with placebo is at or below 0.05. Otherwise, the lower of the two p values must be at or below 0.025 to allow rejection of the null hypothesis for the representative dose.
Changes from baseline in other endpoints (for each subtest and total) were analyzed in a manner similar to the primary endpoint or using an analysis of covariance (ANCOVA) of ranked data if assumptions of normality were severely violated. The change from baseline in CGI severity and improvement scores were analyzed by means of ANCOVA of ranked data, with treatment (MIN-101 and placebo) as a factor and baseline CGI severity score as a covariate for both endpoints.
Results
A total of 244 patients were randomly assigned to a treatment group (safety population) and received at least one dose of study medication. All patients were Caucasian, and 137 (56%) were male. The median age was 41 years, and the mean BMI was 25.6. A total of 234 patients (96%) were included in the intent-to-treat efficacy population (having at least one on-treatment efficacy evaluation) (see Figure S1 in
data supplement that accompanies the online edition of this article). At baseline, the patients were moderately ill, with a mean total score of 80.3 on the PANSS and a mean score of 26.8 on the PANSS “classic” negative subscale (items N1–N7), well above the entry criterion threshold of 20 points. The three treatment groups were balanced on all demographic and illness-related baseline characteristics and prior pharmacological treatment. Nearly 70% of the patients were treated with oral second-generation antipsychotics before entering the trial (
Tables 1 and
2).
Efficacy
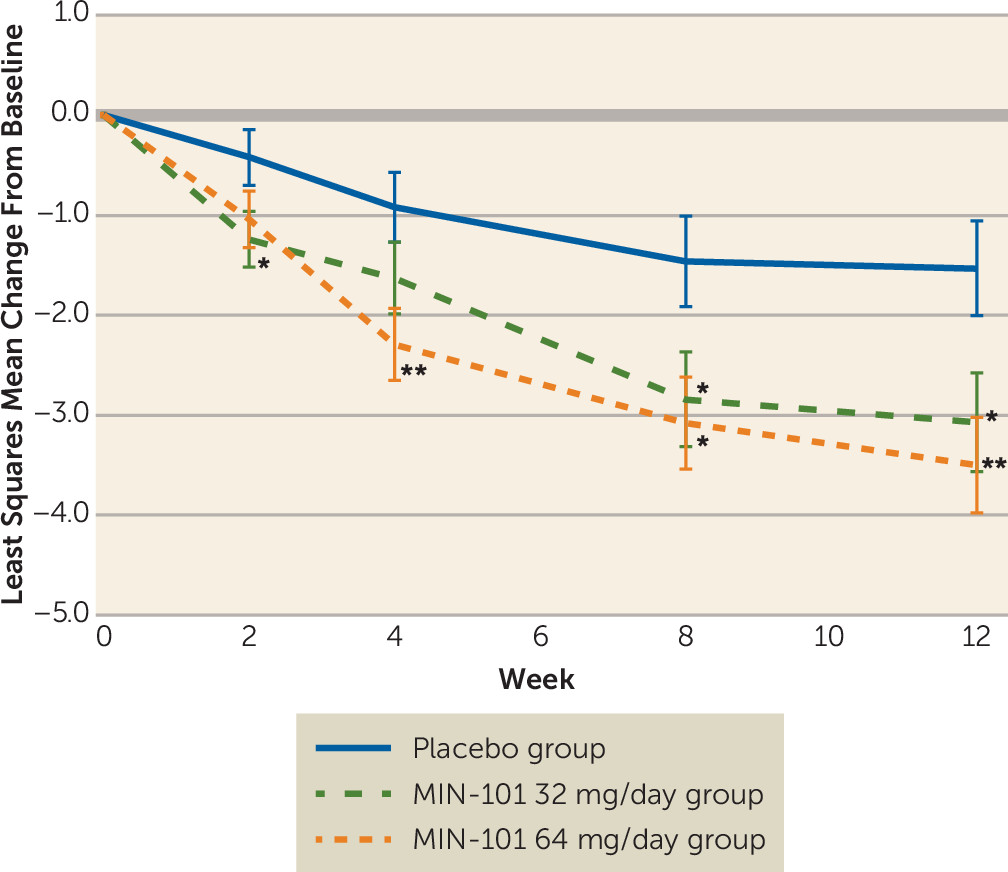
At the end of 12 weeks, there was a statistically significant reduction in the primary endpoint, the PANSS negative symptom pentagonal structure factor score (N1–N4, G5–G8, G13, G14), for the MIN-101 32 mg/day and 64 mg/day groups compared with the placebo group (p≤0.024, d=0.45, and p≤0.004, d=0.57, respectively) (
Figure 1). Statistically significant improvements were seen for the MIN-101 32 mg/day group at 2 weeks and for both MIN-101 dosage groups at 8 weeks, with benefit maintained throughout the entire 12-week treatment period (
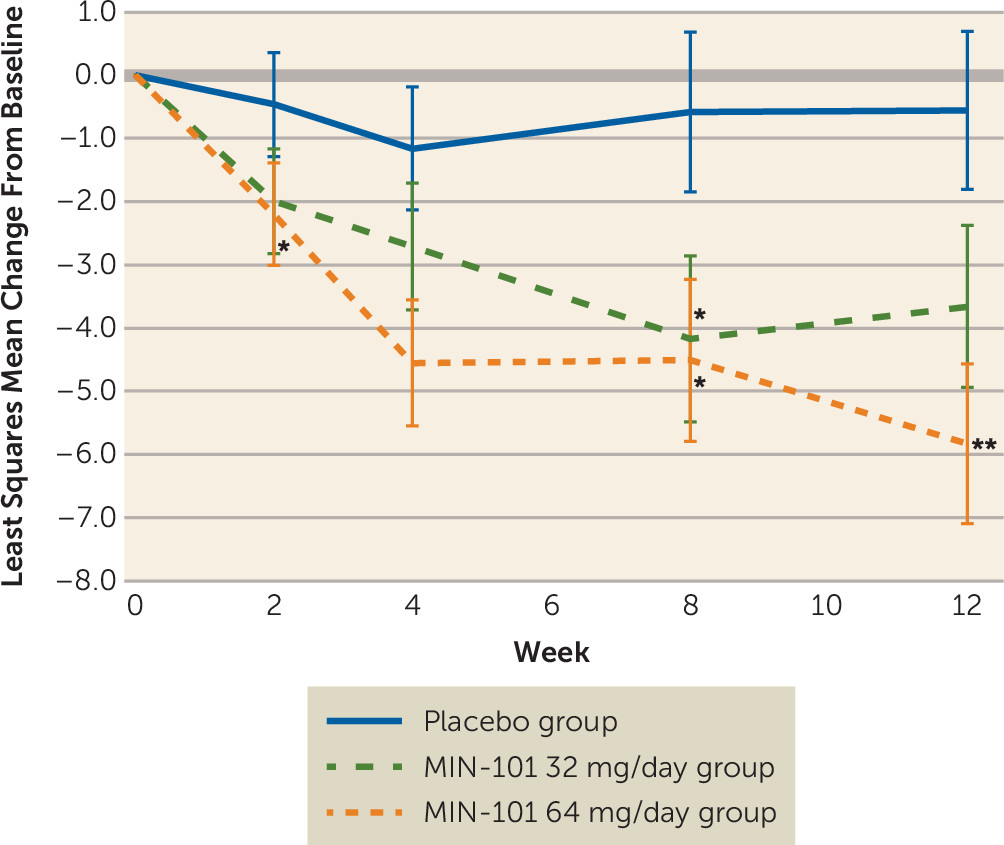
Figure 1). Similar findings were observed on the PANSS negative “classic” (N1–N7) scale (see Figure S2 in the
online data supplement) and PANSS total score (
Figure 2). A per-protocol analysis of the primary outcome measure, the PANSS negative symptom pentagonal structure factor score, also showed a statistically significant reduction for both the MIN-101 32 mg/day group (N=48) and the 64 mg/day group (N=53) compared with the placebo group (N=53), at the end of 12 weeks (p≤0.028, d=0.34, and p≤0.009, d=0.41, respectively). Consistent with these results was the statistically significant superiority of MIN-101 over placebo on PANSS total score and the PANSS five-factor activation score, and, in the 64 mg/day group only, PANSS five-factor and dysphoric mood scores, CGI severity and improvement scores, Brief Negative Symptom Scale score, and CDSS score. The overall composite BACS score for the treatment groups was not significantly better than that for the placebo group (
Table 3).
Because there was a significant change on both negative symptoms and depression, we conducted additional post hoc analyses to investigate the extent to which change in negative symptoms was independent of change in depression. First, we examined the Pearson correlation between change from baseline to endpoint on depression, as measured by the CDSS total score, and change from baseline to endpoint on the negative symptom pentagonal structure factor model, which yielded an r value of 0.26. The low correlation between changes in depression and negative symptoms supports the view that improvement in negative symptoms was not synonymous with improvement in mood.
Second, we performed an ANCOVA to examine treatment effects (change from baseline to endpoint) before and after controlling for changes from baseline to endpoint on the CDSS scores. We found that the superiority of both doses of MIN-101 compared with placebo for the PANSS negative pentagonal factor model was maintained. After controlling for change in depression, the effect size on the PANSS negative pentagonal factor decreased by only 0.03 for the MIN-101 32 mg/day dosage as compared to placebo, and by 0.11 for the 64 mg/day dosage as compared to placebo.
There were no statistically significant differences between the three treatment groups in PANSS positive symptom scores at week 12, nor was there significant worsening compared with baseline. Discontinuation prior to week 12 because of worsening of schizophrenia symptoms or withdrawal of consent (which could be a surrogate for symptom worsening) was reported in 24% of the patients in the placebo group, 18% in the MIN-101 32 mg/day group, and 14% in the MIN-101 64 mg/day group (see Figure S1 in the online data supplement). At least one dose of a benzodiazepine or a hypnotic drug for agitation or insomnia was administered to 25% of patients in the placebo group, 20% of the MIN-101 32 mg/day group, and 23% of the MIN-101 64 mg/day group.
Safety and Tolerability
There was no notable change in body weight from baseline to study end in any of the three groups (placebo group, −0.91 kg [SD=3.30]; MIN-101 32 mg/day group, −0.23 kg [SD=2.10]; MIN-101 64 mg/day group, 0.05 [SD=2.69]), and the three groups had similar decreases in prolactin plasma levels. There were no changes in vital signs, routine laboratory values, and extrapyramidal symptom ratings as measured by AIMS score. There were no changes in suicidality as indicated by score on the Sheehan–Suicidality Tracking Scale. Eight patients experienced serious adverse events (two in the placebo group and six in the MIN-101 groups), of whom six were hospitalized for worsening of schizophrenia symptoms (two in the placebo group and four in the MIN-101 32 mg/day group). The two remaining serious adverse events occurred in the MIN-101 64 mg/day group: vomiting and abdominal pain in one patient and syncope and bradycardia in the other.
During the study, 57.7% of patients in the MIN-101 32 mg/day group, 57.1% of patients in the MIN-101 64 mg/day group, and 43.4% of patients in the placebo group reported at least one treatment-emergent adverse event. The most commonly reported events for patients in the MIN-101 groups were headache (3.6% in the placebo group, 7.5% in the MIN-101 groups), anxiety (6.0% in the placebo group, 6.8% in the MIN-101 groups), insomnia (9.6% in the placebo group, 5.6% in the MIN-101 groups), schizophrenia symptoms (10.8% in the placebo group, 5.6% in the MIN-101 groups), asthenia (2.4% in the placebo group, 5.6% in the MIN-101 groups), nausea (3.6% in the placebo group, 3.7% in the MIN-101 groups), and somnolence (0.0% in the placebo group, 3.7% in the MIN-101 groups). The remainder of reported treatment-emergent adverse events occurred in ≤2.5% of the MIN-101 patients overall.
Anticholinergic drugs, which had been discontinued at baseline, were given to four patients in the placebo group, none in the MIN-101 32 mg/day group, and two in the MIN-101 64 mg/day group. Antianxiety medications were given to nine patients in the placebo group, 10 in the MIN-101 32 mg/day group, and 13 in the MIN-101 64 mg/day group.
Discussion
In this 12-week randomized double-blind placebo-controlled study of symptomatically stable schizophrenia patients with negative symptoms, MIN-101 at a dosage of either 32 mg/day or 64 mg/day resulted in statistically significantly greater improvement in negative symptoms compared with placebo, as demonstrated by the primary outcome measure, change in PANSS negative symptom five-factor pentagonal model score (N1–N4, G5–G8, G13, G14) and on the “classic” (N1–N7) negative symptom scale. Statistically significant improvement for MIN-101 compared with placebo was also observed on certain secondary measures. MIN-101 did not produce the adverse effects generally attributed to antipsychotic drugs, thus reducing the likelihood that patients and raters could have been unblinded by observation of such effects.
A relevant clinical question is whether the improvement in negative symptoms reported here is specific and clinically meaningful. Indeed, it is possible that MIN-101 has a beneficial effect on mood. However, the effect on negative symptoms was maintained after controlling for depression, which suggests the effect was not synonymous with improvement in mood. It is also conceivable that the phenomena measured by negative symptoms and mood scales partially overlap and that a single underlying biological effect of MIN-101 is responsible for the changes in all such scales. It could also be argued that a nonspecific, secondary effect on negative symptoms could be obtained with the help of antipsychotic D
2 blocking drugs. However, examination of negative symptoms in patients treated with antipsychotic drugs in the Novel Methods Leading to New Medications in Depression and Schizophrenia (NEWMEDS) cohort reveals an effect size (Cohen’s d) for PANSS negative symptoms of 0.10 (
34), compared with 0.54 for MIN-101 at 32 mg/day and 0.70 for MIN-101 at 64 mg/day. Furthermore, in the present trial, there was no significant improvement in positive symptoms. Similarly, the AIMS scores were low at baseline and showed only small variations throughout the trial. Hence, the improvement in negative symptoms cannot be attributed to improvements in extrapyramidal symptoms, at least with respect to dyskinetic symptoms.
Because of the early stage of development of MIN-101, it is difficult to weigh the clinical meaningfulness of the results. Yet there was a noticeable and statistically significantly greater change in CGI severity and improvement scores in the MIN-101 64 mg/day group compared with the placebo group. Furthermore, the effect sizes for negative symptoms reported here are higher than those of currently marketed drugs indicated for schizophrenia and of many drugs utilized to treat chronic diseases in general medicine (
35).
Methodologically, the design of trials targeting negative symptoms raises a number of critical issues related to trial population and patient selection criteria, most of which are still being debated by academics and regulators (
36). For example, an ongoing debate is focused on the operationalization of entry criteria for trials targeting negative symptoms such as prominent, predominant, and European Medicines Agency criteria (
37). On the one hand, in order to maximize the likelihood of detecting a real effect and therefore to avoid pseudo effects, it is important to include prospectively assessed symptomatically stable patients who have relatively severe negative symptoms and only mild positive symptoms. Also, it is important to exclude patients who have symptoms that overlap or mimic negative symptoms, such as extrapyramidal symptoms and major depression. Hence, patients who have at least moderate to severe negative symptoms and no more than mildly to moderately severe positive symptoms and/or depressive symptoms and no extrapyramidal symptoms are ideal for such trials. However, with overly stringent criteria (long-term prospective assessment of symptom stability, high negative symptom ratings, low positive symptom ratings, and no depression or extrapyramidal symptoms), a large proportion of schizophrenia patients would be excluded from trials, making the results less clinically relevant, or less generalizable (
2,
38). Therefore, for this trial we included patients with moderate negative symptoms (PANSS negative symptom scores ≥20), which represents a low threshold. The trial design did not limit the overall severity of positive symptoms, and at baseline some patients experienced such symptoms that could be clinically significant. As evident from
Table 2, based on the threshold entry criteria used in this trial, at baseline patients had moderate to severe negative symptoms (mean rating, 26.8; range, 20–38), very mild depressive symptoms (mean CDSS score, 2.1; range, 0–15), and low positive symptom ratings (mean rating, 14.3; range, 7–22), which remained stable or changed very little, as expected from this chronically ill but stable patient population (
39).
A number of rating scales and factors, such as the 16-item Negative Symptom Assessment, have been reviewed and considered as primary and secondary measures (
40). We selected the PANSS because it is the scale most familiar to investigators and clinicians, and it captures most aspects of the illness. The five-factor (pentagonal) negative factor was selected rather than the negative subscale as the primary outcome measure for two reasons: 1) it demonstrated better signal detection in the previous phase 2a trial (
26), and 2) the PANSS negative score contains two items, “stereotyped thinking” and “difficulty in abstract thinking,” that are outside the currently recommended negative symptom domains (
41). We also used the Brief Negative Symptom Scale, an additional scale assessing negative symptoms, because it distinguishes between anticipatory and consummatory anhedonia and separates internal experience from behavior (
29). The duration of the trial, 12 weeks, and the symptom stability duration of 3 months that were required to qualify for entry into the trial were consistent with most similar trials and current recommendations (
36).
The possibility of conducting an add-on study, in which all patients would be given an antipsychotic treatment followed by MIN-101 or placebo, was considered. However, because MIN-101 may have antipsychotic effects in addition to putative anti-negative-symptom effects, the latter would have been difficult to demonstrate if all patients had been concomitantly treated with another antipsychotic drug.
The effect of MIN-101 on symptoms of schizophrenia can probably be attributed to the synergistic effects on 5-HT
2A and sigma-2 pathways. Drugs acting as antagonists at the 5-HT
2A receptors have already demonstrated some antipsychotic effects (
42) that may have been responsible for the relatively low rate of worsening psychotic symptoms during the trial. Although sigma-2 pathways were shown in preclinical studies to benefit behaviors reflecting negative symptoms and to modulate dopamine activity, it is difficult to speculate on the potential mechanism of action. Furthermore, because the sigma-2 receptor has not yet been cloned, there is currently no available positron emission tomography ligand to measure the level of receptor occupancy and target engagement, either in animals or in human subjects.
Among the limitations of this trial are the short washout period and the possibility that some of the observed improvement might be attributable to the withdrawal of antipsychotics and a decrease in secondary negative symptoms. However, the randomization should have mitigated such effects. The AIMS was the only measure of extrapyramidal symptoms in this study. Thus, while changes (improvements) in parkinsonian symptoms should have been similar across treatment groups as a result of randomization, and thus could not explain the differential change in negative symptom scale scores, we lacked objective, numerical evidence to test this. Future studies should use a more sensitive instrument to assess the full spectrum of extrapyramidal symptoms.
In summary, to our knowledge, this study is unique in reporting on a non-D2-receptor-blocking drug with specific therapeutic effects on negative symptoms of schizophrenia. The validity of these results is supported by findings related to the primary and secondary outcome measures and by the lack of potential confounders, such as improvements in extrapyramidal symptoms or positive symptoms and unblinding through the observation of adverse effects.