Hormonal Treatments for Major Depressive Disorder: State of the Art
Abstract
Hypothalamic-Pituitary-Adrenal Axis
Overview of Physiology
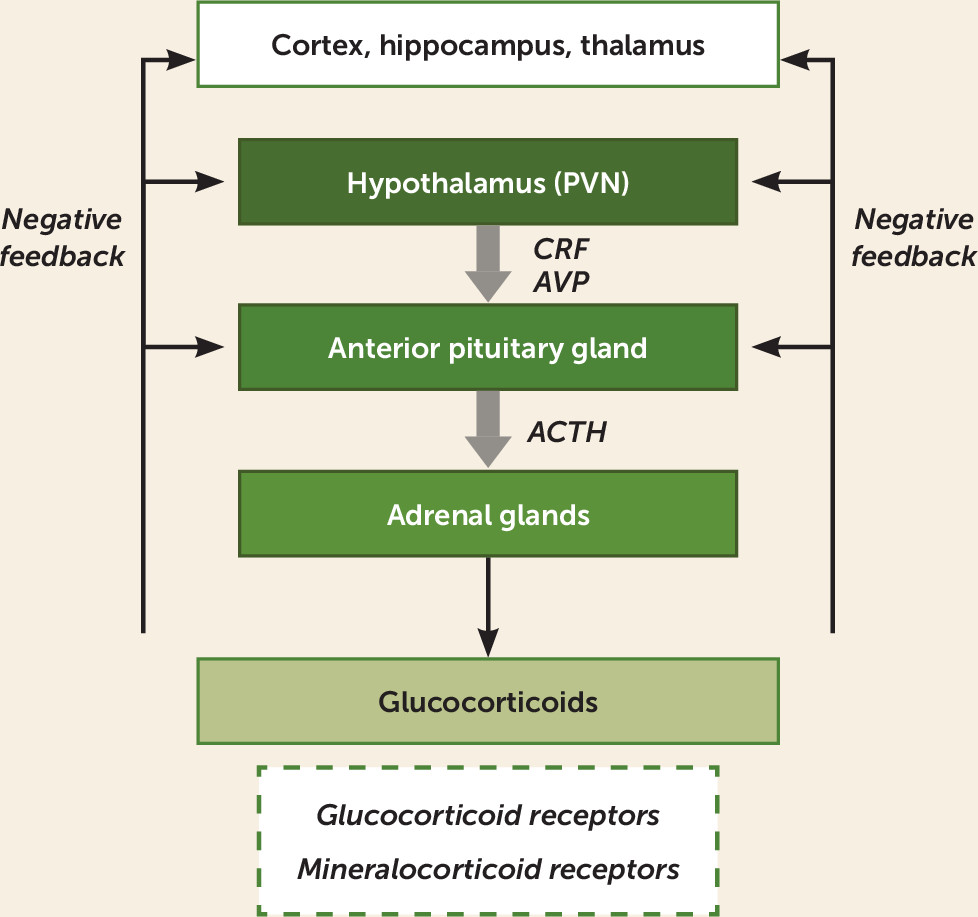
HPA Abnormalities in Major Depression
HPA Axis–Based Treatments for Major Depression
CRFR1 antagonists.
GR antagonists.
Summary of HPA axis–targeted interventions.
| Intervention | Disorder/Population | Level of Evidence |
|---|---|---|
| CRF1 antagonists | Major depression | Strong evidence of no benefit |
| GR antagonists (e.g., mifepristone) | Major depression with psychotic features | Moderate evidence of efficacy for psychotic symptoms if minimum plasma level achieved; additional prospective studies needed |
| T3 augmentation of antidepressants | Treatment-resistant major depression | Moderate evidence of efficacy; efficacy of augmentation with SSRIs requires demonstration in placebo-controlled trials |
| T3 for acceleration of antidepressant effect with TCAs | Major depression | Strong evidence of efficacy |
| T3 for acceleration of antidepressant effect with SSRIs | Major depression | Strong evidence of no benefit |
| Estrogen replacement therapy (or combined hormone replacement therapy) | Perimenopausal women with major depression and physical menopause symptoms | Moderate evidence of efficacy |
| Estrogen replacement therapy (or combined hormone replacement therapy) | Perimenopausal women without major depression (prevention) | Weak evidence of benefit |
| Estrogen replacement therapy (or combined hormone replacement therapy) | Postmenopausal women with major depression | Poor as monotherapy, preliminary evidence as adjunct to SSRIs in geriatric depression |
| Oral contraceptives | PMDD | Moderate evidence of efficacy for drospirenone-containing oral contraceptives, weak evidence of efficacy for other oral contraceptives, despite being considered second-line treatment after SSRIs |
| Allopregnanolone stabilization | PMDD | Moderate evidence of efficacy |
| Allopregnanolone enhancement | Postpartum depression | Strong evidence of efficacy |
| Testosterone replacement therapy | Depressive symptoms secondary to clinical hypogonadism | Strong evidence of efficacy |
| Testosterone augmentation | Subthreshold depressive symptoms in men without clinical hypogonadism | Preliminary evidence of efficacy |
| Testosterone augmentation | Treatment-resistant major depression in men without clinical hypogonadism | Strong evidence of no benefit |
Hypothalamic-Pituitary-Thyroid (HPT) Axis
Overview of Physiology
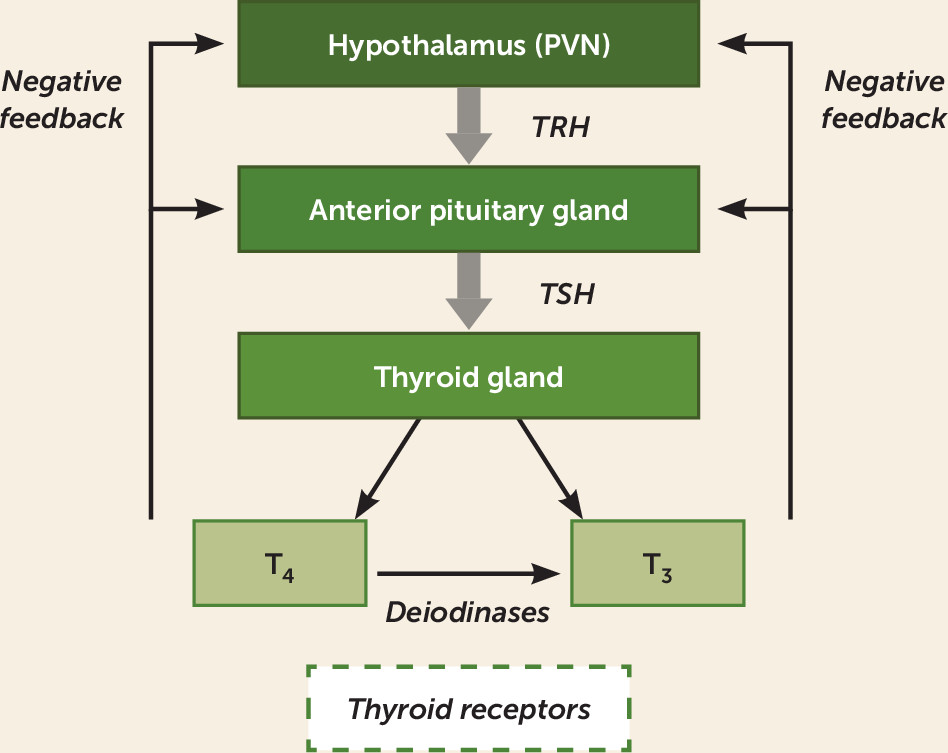
HPT Abnormalities in Major Depression
HPT Axis–Based Treatments for Major Depression
Triiodothyronine (T3).
Acceleration of antidepressant effect with tricyclic antidepressants
Augmentation strategy for inadequate response to antidepressant monotherapy
Thyroxine (T4).
HPT axis: conclusions.
Hypothalamic-Pituitary-Gonadal (HPG) Axis: Ovarian Hormones
Overview of Physiology
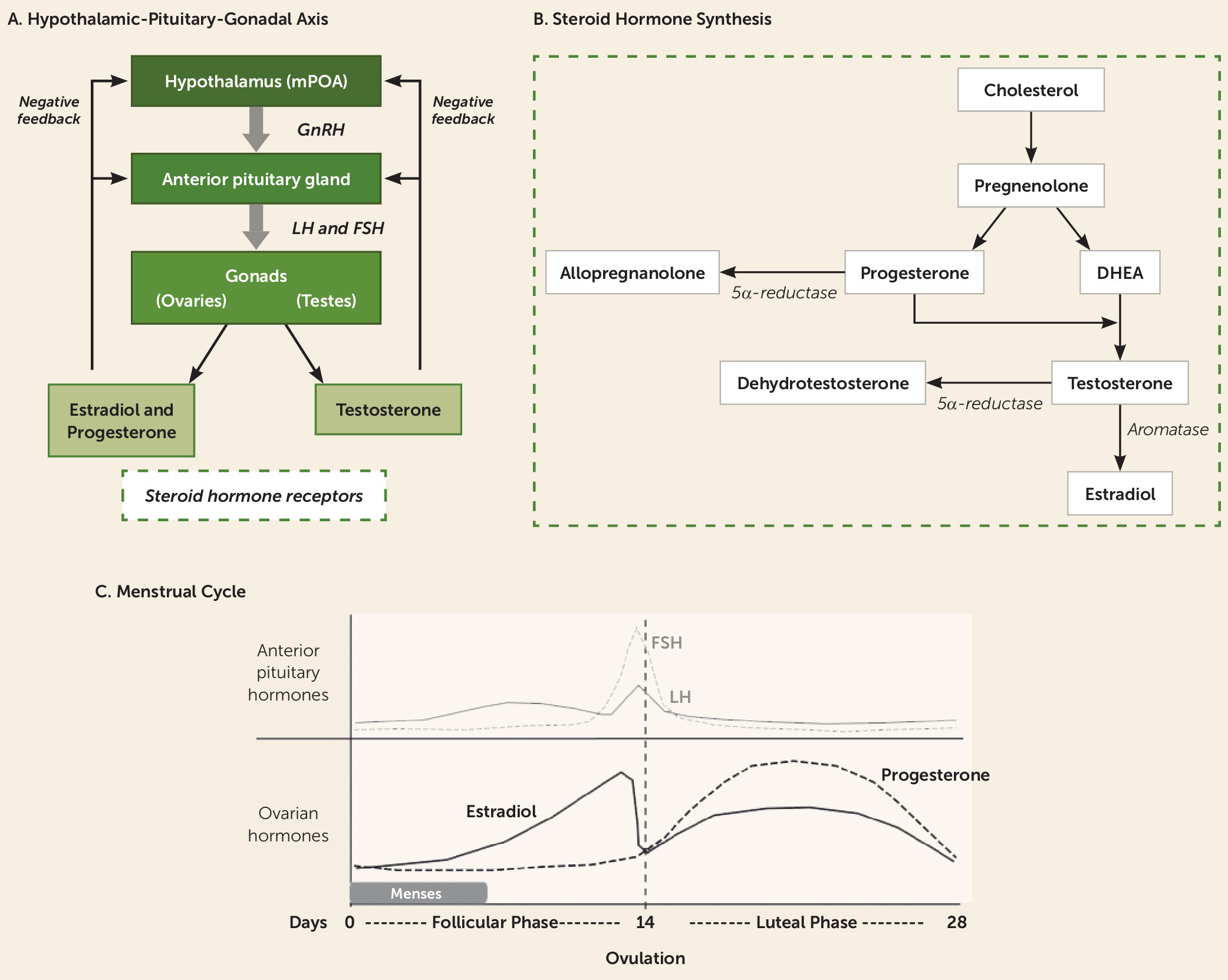
Ovarian Hormone Abnormalities in Major Depression
Ovarian Hormone Treatments for Major Depression
Estrogen replacement therapy (ERT) or hormone replacement therapy (HRT; estrogen and progestin) in peri- or postmenopausal major depression.
Progesterone and its neurosteroid derivative, allopregnanolone.
Ovarian hormone treatment: conclusions.
Hypothalamic-Pituitary-Gonadal (HPG) Axis: Testicular Hormones
Overview of Physiology
Testicular HPG Axis Abnormalities in Major Depression
HPG Axis–Based Treatments for Major Depression
Conclusions and Future Directions
Acknowledgments
Footnote
Supplementary Material
- View/Download
- 394.17 KB
- View/Download
- 82.22 KB
- View/Download
- 199.37 KB
References
Information & Authors
Information
Published In


History
Keywords
Authors
Competing Interests
Funding Information
Metrics & Citations
Metrics
Citations
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
For more information or tips please see 'Downloading to a citation manager' in the Help menu.
View Options
View options
PDF/EPUB
View PDF/EPUBLogin options
Already a subscriber? Access your subscription through your login credentials or your institution for full access to this article.
Personal login Institutional Login Open Athens loginNot a subscriber?
PsychiatryOnline subscription options offer access to the DSM-5-TR® library, books, journals, CME, and patient resources. This all-in-one virtual library provides psychiatrists and mental health professionals with key resources for diagnosis, treatment, research, and professional development.
Need more help? PsychiatryOnline Customer Service may be reached by emailing [email protected] or by calling 800-368-5777 (in the U.S.) or 703-907-7322 (outside the U.S.).