The microbiota can be influenced by many factors, including diet, exercise, and infection, and stress is a major factor that is relevant for depression (
9–
11). Substantial evidence demonstrates that activation of the hypothalamic-pituitary-adrenal (HPA) axis influences the composition of the gut microbiota. Maternal separation in rhesus monkeys, which increases HPA axis activity, was found to result in a reduction of fecal bacteria, especially
Lactobacilli (
12,
13), and early-life stress in rats or chronic restraint stress in adult rodents also changed the composition of the gut microbiota (
14), in conjunction with increasing proinflammatory cytokines and chemokines (
15). Chronic restraint stress also increases the permeability of the gut, leading to the disruption of the gut barrier (leaky gut) and the release of lipopolysaccharides into the circulation (
16,
17). This can be reversed, however, by probiotic agents (
13,
17,
18). Patients with major depression exhibit significant changes in the relative abundance of Firmicutes, Actinobacteria, and Bacteroidetes compared with healthy individuals (
8), and administration of the probiotic
Lactobacillus farciminis has been shown to prevent the gut leakage and the activation of the HPA axis (
19,
20), confirming the critical role of the microbiota in the gut-brain dialogue and the potential beneficial role of probiotic treatments in major depression (
21). A variety of mechanisms of action of probiotics have been proposed, such as 1) down-regulating the HPA axis, which is often overactive in patients with major depression (
22); 2) promoting biosynthesis of GABA, levels of which are reduced in patients with major depression (
23); and 3) increasing the production of tryptophan and therefore the serotonin level (
24). Altogether, gut dysbiosis is often evident in patients with major depression and in mice exhibiting depressive-like behaviors (
8,
25–
29; for a review, see reference
30), suggesting a dysregulation of the brain-gut axis in depression. However, molecular mediators of the effects of the microbiota in depressive-like behaviors remain to be identified.
Besides regulating brain responses to stress and behavior, the microbiota also dramatically regulates the immune system (
31,
32). Recent studies have shown the importance of host-specific species such as the segmented filamentous bacteria (SFB), also called “
Candidatus Arthromitus.” SFB is a nonculturable spore-forming Gram-positive bacterium that is most closely related to the genus
Clostridium and colonizes the intestines of numerous species, including humans (
33). Mouse gut colonization by SFB is required for the maturation of the innate and the adaptive immune systems (
34,
35). This is relevant for depression because critical roles of the innate and adaptive immune systems have been shown to regulate the susceptibility to depressive-like behaviors in mice (
36–
38). T cells express the surface marker, CD4, that is used to identify them as CD4
+, and one type of T cell, the T helper (Th) 17 cell that produces the signature cytokine interleukin 17A (IL-17A), is known to be toxic to the CNS in autoimmune diseases (
39). We recently found that Th17 cells were sufficient to increase susceptibility to depressive-like behaviors in mice (
40). Th17 cells are only present in the lamina propria of the small intestine in healthy mice (
34), and they are pathogenic when they infiltrate the CNS, such as in multiple sclerosis (
41). Thus, it is particularly relevant that Th17 cells are highly up-regulated by SFB (
34,
42). Without SFB, Th17 cells are absent in the mouse small intestinal lamina propria, where they are usually abundant in normal healthy rodents in the presence of commensal microbiota (
34). Introduction of SFB in mice that are deficient in Th17 cells induces the production of Th17 cells (
34).
Methods
Mice
We used 6- to 12-week-old wild-type C57BL/6, Rorc(γT)
+/GFP, Rag2
−/−, or 7B8 (Tg(Tcra,Tcrb)2Litt/J, strain 027230), IL17A-IRES-GFP-KI (strain 018472) male mice. C57BL/6 mice were either bred in the University of Miami animal facility or purchased from Taconic Biosciences (TAC) or Jackson Laboratory (JAX). The Rorc(γT)
+/GFP mice (strain 007572 [
44]) were obtained by crossing Rorc(γT)
+/GFP × Rorc(γT)
+/GFP to produce 50% Rorc(γT)
+/GFP, 25% wild-type, and 25% Rorc(γT)
GFP/GFP mice, and littermates were used. Rorc(γT)
GFP/GFP mice were not used because their locomotor activity is altered, compromising the interpretation of the behavioral testing. Mice were housed in light- and temperature-controlled rooms and treated in accordance with National Institutes of Health and the University of Miami Institutional Animal Care and Use Committee regulations.
Treatments
Mice were injected intraperitoneally with 5 nmol of AI-2 (Omm Scientific, Dallas; vehicle is saline), short-chain acyl homoserine lactone (sc-AHL) (chain of 6 carbons) (Sigma-Aldrich, St. Louis; vehicle is saline), or oleic acid (Sigma-Aldrich, St. Louis; vehicle is saline with 5% DMSO, 5% Tween80), or 5 μg of mouse recombinant SAA2 (MyBioSource, San Diego; vehicle is saline), as indicated within each experiment.
Behavioral Assessments
Learned Helplessness.
Learned helplessness was measured using a standard learned helplessness paradigm or a modified reduced inescapable foot shock protocol, as described previously (
40,
45). The reduced paradigm was used so that wild-type C57BL/6 mice did not develop learned helplessness, allowing measurements of increased susceptibility to learned helplessness. Briefly, mice were placed in one side of a Gemini avoidance system shuttle box (Med Associates, St. Albans, Vt.) with the gate between chambers closed. For standard learned helplessness, 180 inescapable foot shocks were delivered at an amplitude of 0.3 mA, a duration of 6–10 seconds per shock, and a randomized intershock interval of 5–45 seconds (
40). In a modified inescapable shock protocol, referred to as the reduced learned helplessness protocol, mice were given 180 foot shocks with amplitude of 0.3 mA and a 2- to 6-second shock duration, and a randomized intershock interval of 5–45 seconds (
45). Twenty-four hours after the inescapable foot shocks, mice were returned to the shuttle box and the number of escapes from 30 escape trials was recorded. Each trial uses a 0.3 mA foot shock for a maximum duration of 24 seconds. The door of the chamber opens at the beginning of the foot shock administration to allow the mouse to escape. Trials in which the mouse did not escape within the 24-second time limit were counted as escape failures. Mice with >15 escape failures were defined as learned helpless. For the long-term paradigm, mice subjected to the standard learned helplessness were retested with escapable foot shocks once a week over a maximum of 4 weeks (
46).
Tail Suspension Test.
For the tail suspension test, mice were suspended by the tail in an automated tail suspension test cubicle (33×31.75×33 cm; Med Associates) for a period of 6 minutes, and the immobile time was analyzed for the last 4 minutes, using Med Associates software.
Open Field.
Locomotor activity in an open field apparatus was measured as previously described (
40). Briefly, mice were placed in a Plexiglas open field (San Diego Instruments, San Diego) outfitted with photobeam detectors under soft overhead lighting, and activity was monitored over 30 minutes using activity monitoring software (San Diego Instruments).
Social Interactions.
For the three-chambered social interaction test (
40), the apparatus was a transparent rectangular Plexiglas box divided by Plexiglas walls into three equal-sized connected chambers with an empty wire enclosure in the two end chambers. The day before testing, the test mice were habituated individually by being allowed to freely explore the entire apparatus for 20 minutes and, separately, an unfamiliar, conspecific, same-sex stimulus mouse was habituated for 20 minutes in the wire enclosure in one of the chambers. On the day of the test, the test mouse was placed in the center of the middle chamber and allowed to freely explore the entire apparatus for 5 minutes. The test mouse was allowed to explore the entire apparatus for 10 minutes with the unfamiliar mouse placed in one of the chambers on the side of the box. Each session was videotaped and quantified for time spent in each chamber and for number of nose contacts with the stimulus mouse.
SFB Colonization
The colonization by SFB was achieved by either ad libitum cohousing 5–6 week old age-matched male mice in sterilized cages for 2 weeks at a ratio of 2:3 JAX to TAC mice, as previously described (
47), or by gavage of 100 μL of fecal homogenates of SFB monocolonized mice in water (
34). SFB colonization after 2 weeks was confirmed by quantitative polymerase chain reaction (qPCR). Control JAX mice were gavaged with homogenates of their own feces in water.
SFB qPCR
Genomic DNA was purified from stools using the Quick-DNA Fecal/Soil Microbe Miniprep (Zymo Research, Irvine, Calif.) according to the manufacturer’s instructions. SFB gene expression (SFB primers: 5′-ACGCTACATCGTCTTATCTTCCCGC-3′ and 5′-TCCCCCAAGACCAAGTTCACG-3′) was assessed by SYBR qPCR in a qTOWER instrument from Analytik Jena and the results were quantified by the 2−ΔΔCt method. Values were normalized to Eubacteria (EUB primers: 5′-ACTCCTACGGGAGGCAGCAGT-3′ and 5′-ATTACCGCGGCTGCTGGC-3′) for each sample.
Quorum Sensing Molecule Measurement
Fecal homogenates were incubated with bacteria expressing the plasmids pSB406 (sc-AHL), pSB1075 (long-chain acyl homoserine lactone, lc-AHL), or MM32 (AI-2). The same amount of feces weight was used. Briefly, for measuring AHLs, E. coli DH5a bacteria expressing pSB1075 or pSB406 were grown in lysogeny broth (LB) containing 100 μg/mL ampicillin at 37°C with orbital shaking, and for measuring AI-2, Vibrio Harveyi bacteria were grown overnight in M9 medium containing 30 μg/mL kanamycin, at 30°C with orbital shaking (250 rpm). Stools were homogenized in water and diluted 1:1600 (AHL) or 1:750 (AI-2) and incubated for 2 hours (AHLs) or 2.5 hours (AI-2) with these bacteria at 37°C (AHL) or 30°C (AI-2), with orbital shaking at 175 rpm. Bioluminescence was assessed using a standard curve with commercial AHL or AI-2 (Sigma-Aldrich or Omm Scientific). The induced luminescence intensity was measured using a microplate reader (BMG Labtech Polar Star Optima microplate luminometer, Ortenberg, Germany). All luminescent intensities were reported as the average of a minimum of three replicates that are blank subtracted and are expressed in relative light units.
Microbiome Sequencing
V4 16S RNA sequencing was performed by Second Genome (South San Francisco, Calif.). Briefly, nucleic acid isolation was achieved with the MoBio PowerMag Microbiome kit (Mo Bio Laboratories, Carlsbad, Calif.) according to the manufacturer’s instructions and optimized for high-throughput processing. All samples were quantified via the Qubit Quant-iT dsDNA High Sensitivity Kit (Invitrogen, Life Technologies, Grand Island, N.Y.).
For the library preparation, each sample was PCR amplified with two differently bar-coded V4 fusion primers and concentrated using a solid-phase reversible immobilization method for the purification of PCR products and quantified by qPCR. Samples were paired-end sequenced using a MiSeq instrument (Illumina, San Diego) according to the manufacturer’s instructions. Reads were merged using USEARCH, and the resulting sequences were compared with an in-house strains database using USEARCH (usearch_global). All sequences hitting a unique strain with an identity ≥99% were assigned a strain operation taxonomic unit (OTU). To ensure specificity of the strain hits, a difference of ≥0.25% between the identity of the best hit and the second best hit was required. For each strain OTU, one of the matching reads was selected as representative and all sequences were mapped by USEARCH (usearch_global) against the strain OTU representatives to calculate strain abundances. The remaining non-strain sequences were quality filtered and dereplicated with USEARCH. Resulting unique sequences were then clustered at 97% by UPARSE, and a representative consensus sequence per de novo OTU was determined. Representative OTU sequences were assigned taxonomic classification via the mothur Bayesian classifier, trained against the Greengenes reference database of 16S rRNA gene sequences clustered at 99%. All profiles are intercompared in a pairwise fashion to determine a dissimilarity score and stored in a distance dissimilarity matrix, using the Bray-Curtis dissimilarity. The binary dissimilarity values were calculated with the Jaccard index and represented using dendrograms and analyzed using permutational analysis of variance. Univariate differential abundance of OTUs was tested using a negative binomial noise model for the overdispersion and Poisson process intrinsic to these data, as implemented in the DESeq2 package (
48,
49). DESeq q-values were corrected using the Benjamini-Hochberg procedure. All the data were deposited in the Sequence Read Archive (SRA), Bioproject PRJNA498103.
Antibiotic Treatment
Specific-pathogen-free (SPF) mice were gavaged with a solution of neomycin (100 mg/kg), metronidazole (100 mg/kg), and vancomycin (50 mg/kg) twice daily for 7 days before starting the injections of AI-2, and antibiotic treatments were continued until the behavioral assessments were finished to avoid bacterial recolonization, using dosages typically greater than those used for clinical therapeutic treatment. Ampicillin (1 mg/mL) was also provided ad libitum in drinking water. These conditions produced germ-free-like phenotype (
50). Bacterial depletion was evaluated by
Eubacteria 16S qPCR. The level of
Eubacteria was 99.9% depleted (see Figure S1F in the
online supplement), confirming depletion of the microbiota with the antibiotic regimen.
Adoptive Transfer
CD4
+ cells were isolated as described previously (
33). Approximately 1×10
6 undifferentiated CD4 cells were injected intravenously in 200 μL phosphate-buffered saline (PBS) by tail vein 48 hours before behavioral testing or as indicated in Figure S2H–I in the
online supplement. Depending on the number of mice to be injected, cells transferred were pooled from 1–2 donors.
SAA, IL-22 Quantitative Real-Time PCR (qRT-PCR)
The most distal part of the small intestine was dissected and rinsed and Peyer’s patches were removed. RNA was extracted with TRIzol Reagent (Life Technologies) and cDNA was synthesized with ImProm-II Reverse Transcriptase and random primers (Promega, Madison, Wisc.). SAA1, SAA2, SAA3 (
34), and IL-22 expression was measured by SYBR green RT-qPCR in a qTOWER instrument from Analytik Jena and the results were quantified by the 2
−ΔΔCt method. The primers used were SAA1: 5′-CATTTGTTCACGAGGCTTTCC-3′ and 5′-GTTTTTCCAGTTAGCTTCCTTCATGT-3′; SAA2: 5′-TGTGTATCCCACAAG GTTTCAGA-3′ and 5′-TTATTACCCTCTCCTCCTCAAGCA-3′; SAA3: 5′-CGCAGCACGAGCAGGAT-3′ and 5′-CCAGGATCAAGATGCAAAGAATG-3′; IL-22: 5′-AGAACGTCTTCCAGGGTGAA-3′ and 5′-TCCGAGGAGTCAGTGCTAAA-3′. Values were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (primers: 5′-AGGTCGGTGTGAACGGATTTG-3′ and 5′-TGTAGACCATGTAGTTGAGGTCA-3′).
Flow Cytometry
Immediately after learned helplessness, mice were anesthetized, spleens were recovered, and mice were transcardially perfused with PBS as previously described (
40,
45). Briefly, the hippocampi were dissected, excluding meninges and choroid plexus, passed through a 70-μm cell strainer (BD Biosciences, San Jose, Calif.), and the cell suspension was mixed (vol/vol) to obtain a 30% Percoll/R1 medium (RPMI 1640 medium [Corning] supplemented with 1% fetal bovine serum [Gibco], 100 IU/mL penicillin [Gibco], 100 μg/mL streptomycin [Gibco], 1×nonessential amino acids [Gibco], 1 μM sodium pyruvate [Gibco], 2.5 μM β-mercaptoethanol [Sigma-Aldrich] and 2 mM
l-glutamine [Gibco]). The cellular suspension was overlaid on 70% Percoll/R1 medium in a centrifuge tube and centrifuged at 2000 rpm for 20 minutes without using the brake. The cells at the interface of the 30%/70% Percoll gradient were recovered, washed once, resuspended in R10 media and stimulated for 4 hours with phorbol myristate acetate (50 ng/mL; Sigma-Aldrich) and ionomycin (750 ng/mL; Sigma-Aldrich) in the presence of Protein Transport Inhibitor Cocktail (eBioscience) at the recommended concentrations. Standard intracellular cytokine staining was carried out as described elsewhere (
33) using the Staining Intracellular Antigens for Flow Cytometry Protocol (eBioscience). Cells were first stained extracellularly with BV480-conjugated anti-CD4 (BD Biosciences), then fixed and permeabilized with permeabilization solution (eBioscience), and finally intracellularly stained with BV650-conjugated anti-IFN-γ (BD Biosciences), phycoerythrin-conjugated anti-IL-17A (eBioscience) and BV421-conjugated anti-FoxP3 (BD Bioscience). Samples were acquired on a FACS CELESTA (BD Biosciences) and data were analyzed with the FlowJo software program (Tree Star, Inc.).
Immunohistochemistry
Immediately after learned helplessness escapable-shock testing, mice were anesthetized, spleens were recovered, and mice were transcardially perfused with PBS as described above. Brains were fixed with 4% PFA, postfixed, cryo-protected in sucrose, and then snap-frozen in optimal cutting temperature reagent and stored at −80°C. Twenty-micrometer sections were stained for GFP (anti-GFP Chicken, catalog number 1020, Aves Labs) and Dapi and imaged on an EVOS FL fluorescent microscope (Life Technologies).
Gut Permeability
To measure gut permeability (modified from reference
51), mice were gavaged with FITC-Dextran (Sigma-Aldrich; 600 mg/kg in 100 μL of PBS) 4 hours before sacrifice. Mice were anesthetized and blood was collected at the time of sacrifice via hepatic vein and spun down to collect serum. Fluorescence was measured at 490 nm (excitation) and 530 nm (emission) in serum samples diluted 1:1 with PBS.
Cytokine Measurements
Serum cytokines were measured in 12.5 μL of serum using multiplexing measuring 26 cytokines and chemokines according to the manufacturer’s instructions (mouse cytokine/chemokine, 26 Plex, catalog number EPX260-26088-901, eBioscience) on a MAGPIX instrument (Luminex).
Human Samples
Stool samples from 10 participants with current depressive symptoms, defined as a score ≥13 on the Quick Inventory of Depressive Symptomatology (QIDS), and 10 matched healthy control subjects were analyzed (for demographic information, see in Table S1 in the online supplement) from an ongoing longitudinal study at the University of Texas Southwestern Medical Center. Samples were collected by participants at home, frozen upon collection, transported frozen back to the center, and then stored at −80°C until analysis.
Eighty percent of the depressed samples had a primary diagnosis of major depressive disorder, and the remaining psychiatric samples had bipolar disorder diagnoses. Psychiatric state at the time of sample collection, assessed by the Longitudinal Interval Follow-Up Evaluation (LIFE) (
52), confirmed the incidence of a full depressive episode or an episode in partial remission for all. In addition, the majority of the psychiatric samples had marked or fully expressed generalized anxiety disorder or other anxiety disorders, and two participants experienced mania or hypomania within 2 weeks of sample collection. One participant’s LIFE data were not available. The healthy control samples had no diagnosed psychiatric disorders.
The study was approved by the University of Texas Southwestern Institutional Review Board. Informed consent was obtained from all participants.
Human IL-17A and SAA Measurement
Stool samples were homogenized (100 mg/mL) in PBS supplemented with 1 μg/mL leupeptin, pepstatin, and 1 mM phenylmethylsulfonyl fluoride, sonicated and centrifuged for 15 minutes at 10,000 g at 4°C. IL-17A and SAA were measured in 25 μL of stool homogenates using human IL-17A Simplex and SAA Simplex Procarta multiplex assays (catalog numbers EXP01A-12136–901, EXP01A-12017–901, Thermo Fisher Scientific) on a MAGPIX instrument (Luminex). Samples were run on one plate, blind to treatment. Assays were checked for quality control to fit the standard curves. Two samples (one from a healthy control subject, one from a participant with major depression who had a primary bipolar diagnosis) were not used for IL-17A and SAA measures because of the nature of the stools (diarrhea), for which 50 mg of actual stool could not be sampled to get protein concentrations consistent with the other samples. This issue did not affect quorum-sensing molecule measurements because they are chemicals measured on the basis of tissue weight rather than protein concentration. Points were excluded if the values deviated more than three standard deviations from the mean and were considered statistically outlier values. For all analyses, any samples that were under the detection limit were excluded. The data were analyzed in SPSS, version 24 (IBM, Armonk, N.Y.).
Human SFB and QSM Measurement
For SFB measurements, genomic DNA was extracted using the Quick-DNA Fecal/Soil Microbe Miniprep (Zymo Research) according to the manufacturer’s instructions, and amplified by PCR as described above. Points were excluded if the values deviated more than three standard deviations from the mean and were considered outlier values. For the QSM measurements, similar approaches to those for mouse were used, as described above.
Statistical Analysis
Data are represented as means and standard errors of the mean. Statistical significance was analyzed with a one-way analysis of variance for multiple comparisons with the Bonferroni post hoc test or with Student’s t test or the Mann-Whitney U test using the Prism program when appropriate. Pearson correlations were computed in SPSS, version 24. The threshold for statistical significance was set at a p value of 0.05. All statistical tests were two-sided.
Results
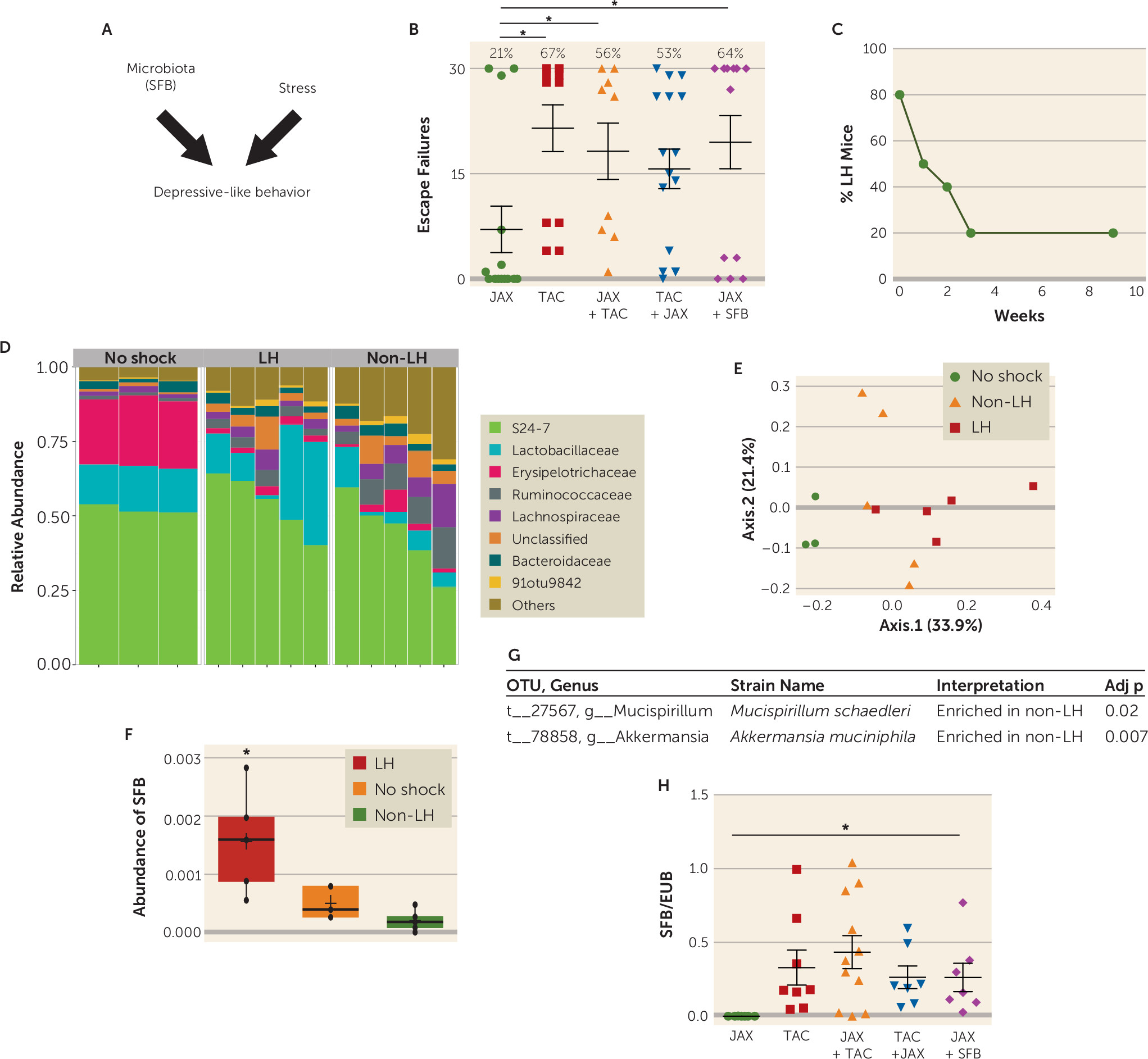
To determine whether the composition of the gut microbiome can affect sensitivity to the foot shock–induced learned helplessness model of depression (
Figure 1A), we tested the sensitivity to the induction of learned helplessness of mice obtained from the Jackson Laboratory (JAX) or Taconic Farms (TAC), which have different microbiomes (
34). We found that only 21% of JAX mice, but 67% of TAC mice, exhibited learned helplessness (
Figure 1B), showing that TAC mice were significantly more susceptible to the induction of learned helplessness than JAX mice. We next tested whether cohousing JAX and TAC mice, which allows sharing of microbiota between cohoused mice, changed their susceptibility to learned helplessness. After cohabitation for 15 days, 56% of JAX mice and 53% of TAC mice exhibited learned helplessness (
Figure 1B). This suggests that JAX mice that acquire the microbiota of TAC mice (
34) display increased susceptibility to learned helplessness compared with naive JAX mice, averaging 18 failures out of the 30 escape trials, compared with eight failures without cohousing, whereas the susceptibility of TAC mice remained essentially unchanged by cohabitation. This raises the possibility that bacteria in the TAC mice microbiota increase sensitivity to learned helplessness, and/or the microbiota of JAX mice provides resilience to learned helplessness.
To identify bacteria that may modulate learned helplessness, TAC mice were retested every week to identify mice that maintained learned helplessness for a long period of time (
Figure 1C). For this, we used the paradigm of learned helplessness we described previously (
40), in which ∼20% of the mice keep exhibiting learned helplessness after 3–9 weeks of retesting (
Figure 1C). This differs from the behavior of most mice, as ∼80% of mice spontaneously recover from learned helplessness within 3 weeks, demonstrating a difference in 20% of mice that blocks recovery from this depression-like behavior. We performed V4 16S rRNA sequencing to identify the bacteria that are different in mouse stools immediately after exposure to escapable foot shocks (to differentiate mice that are either susceptible or resilient to learned helplessness) and in mice that maintain learned helplessness for 4 weeks (
Figure 1D–E). We ensured that a complete microbiome profile was captured because the number of sequences per sample ranged from 111,769 to 162,600 filtered reads. We found no change in the alpha diversity metric, and the average number of OTUs per sample and per group ranged from 342 to 368, with an average Shannon diversity index per group range of 2.77–3.47. There were no differences among groups at the level of phyla. However, at the family level, Erysipelotrichaceae was lower in mice 4 weeks after receiving inescapable foot shocks whether they exhibited (2.26%) or did not exhibit learned helplessness (2.89%) than in mice that did not receive foot shocks (mean 22.7%) (
Figure 1D). Thus, the stress of the learned helplessness paradigm was sufficient to cause this large difference in the microbiota independently of the behavioral outcome. There were six significantly different OTUs detected, out of 497 examined in mice displaying learned helplessness for 4 weeks compared with mice that underwent the learned helplessness paradigm but never displayed learned helplessness.
Akkermansia muciniphila (one OTU) and
Mucispirillum schaedleri (two OTUs) were enriched in non–learned helpless samples (
Figure 1G), whereas SFB (one OTU) and two unclassified OTUs from the order Clostridiales were enriched in mice that displayed learned helplessness for 4 weeks (
Figure 1F). Thus, the microbiome is significantly different in mice that are resilient to learned helplessness and mice that are susceptible to and maintain prolonged learned helplessness.
The elevated SFB level in mice that exhibited prolonged learned helplessness compared with resilient mice raised the possibility that SFB may promote learned helplessness. Interestingly, JAX mice, which are resistant to learned helplessness, are known to be deficient in SFB (
Figure 1H) (
34), and we found that JAX mice cohoused with TAC mice express fecal SFB similar to TAC mice (
Figure 1H) as well as exhibiting similar susceptibility to learned helplessness (
Figure 1B). We tested whether SFB is sufficient to increase susceptibility to learned helplessness by gavaging JAX mice with SFB-monocolonized feces and subjecting them to learned helplessness. After the introduction of SFB, 64% of JAX mice exhibited learned helplessness (compared with 21% in control JAX mice), within an average of 20 failures out of 30 trials (
Figure 1B). These results indicate that the presence of SFB in the microbiota promotes susceptibility to learned helplessness, but they do not rule out the likelihood that other microbiota components also are modulatory, such as
Akkermansia muciniphila and
Mucispirillum schaedleri.
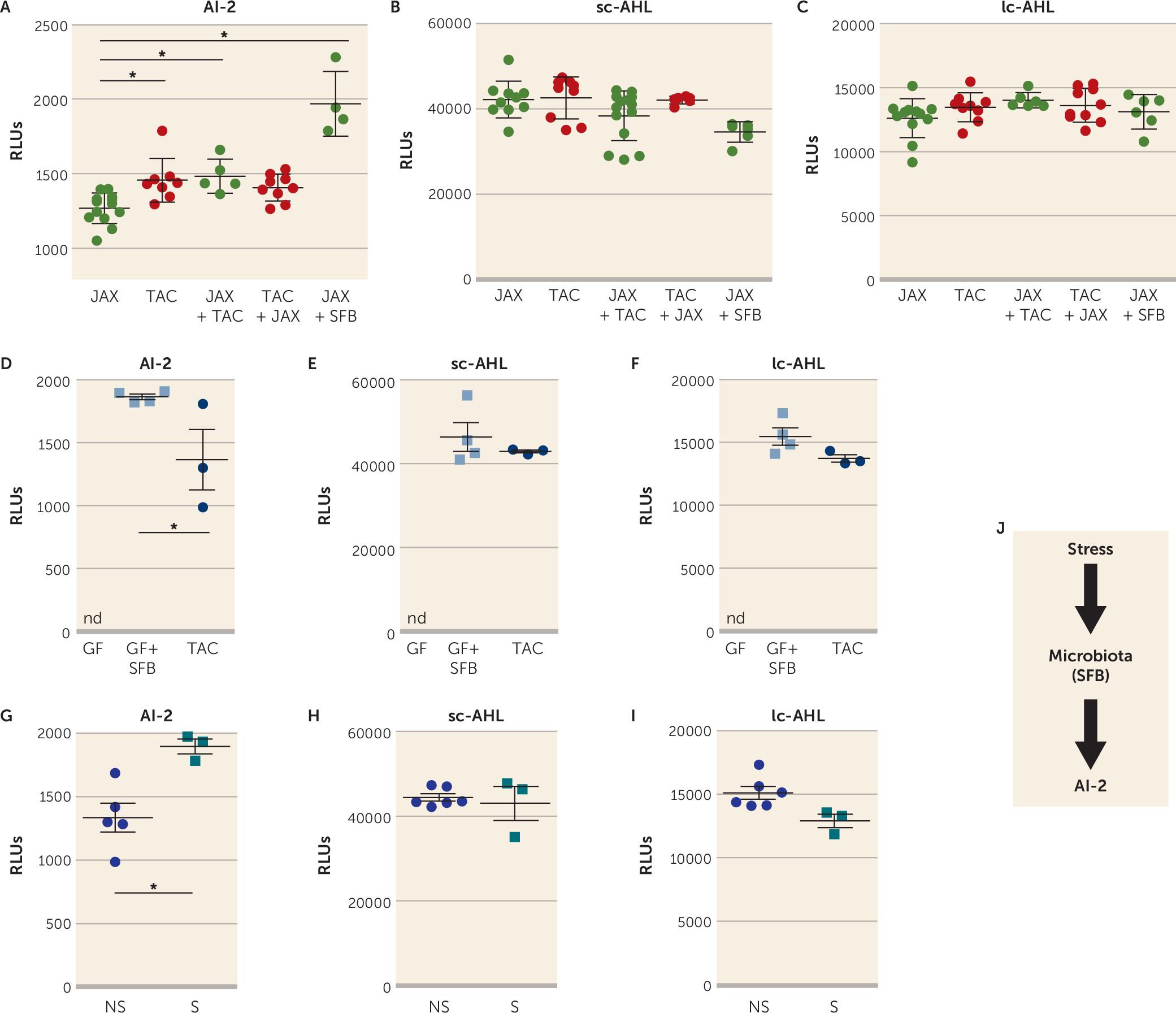
To identify a mechanism by which bacteria in the microbiome may influence depression-like behaviors, we examined quorum-sensing molecules (QSMs). QSMs are uniquely secreted by bacteria in response to changes in population density to coordinate several bacterial behaviors, including biofilm formation, swarming, and virulence gene expression, as well as to modulate antibiotic resistance (
53,
54). We hypothesized that QSMs, which would reflect overall changes in bacteria species in the gut, contribute to microbiome-modulated susceptibility to learned helplessness. We analyzed three major QSMs in the feces of the JAX, TAC, and cohoused mice: autoinducer-2 (AI-2), short-chain AHL (sc-AHL), and long-chain AHL (lc-AHL). The fecal AI-2 level was significantly increased in TAC mice or JAX mice cohoused with TAC mice compared with JAX mice alone (
Figure 2A). The abundance of sc-AHL and lc-AHL was similar among all groups (
Figure 2B-C). Reintroduction of SFB in JAX mice was sufficient to significantly increase AI-2 levels but not sc-AHL or lc-AHL (
Figure 2A–C). We confirmed that AI-2 can be produced by SFB, as the fecal AI-2 level was increased in germ-free mice monocolonized with SFB compared with TAC mice (
Figure 2D), whereas sc-AHL or lc-AHL levels were similar in these two groups (
Figure 2E-F), suggesting that AI-2, sc-AHL, and lc-AHL were all produced by SFB, but that AI-2 might be preferentially expressed in SFB monocolonized mice (
Figure 2D). Germ-free mice do not produce AI-2, sc-AHL, or lc-AHL (
Figure 2D-F). We also determined whether foot shock stress used for inducing learned helplessness induced AI-2 levels. TAC mice subjected to the 48-hour paradigm of learned helplessness had a higher level of AI-2 than TAC mice that did not receive foot shocks (
Figure 2G), whereas the levels of sc-AHL and lc-AHL remained unchanged between these two groups (
Figure 2H–I), indicating that the stress of foot shocks was sufficient to increase AI-2 levels as well as to induce learned helplessness, suggesting that bacteria might adapt their behavior after the induction of stress (
Figure 2J).
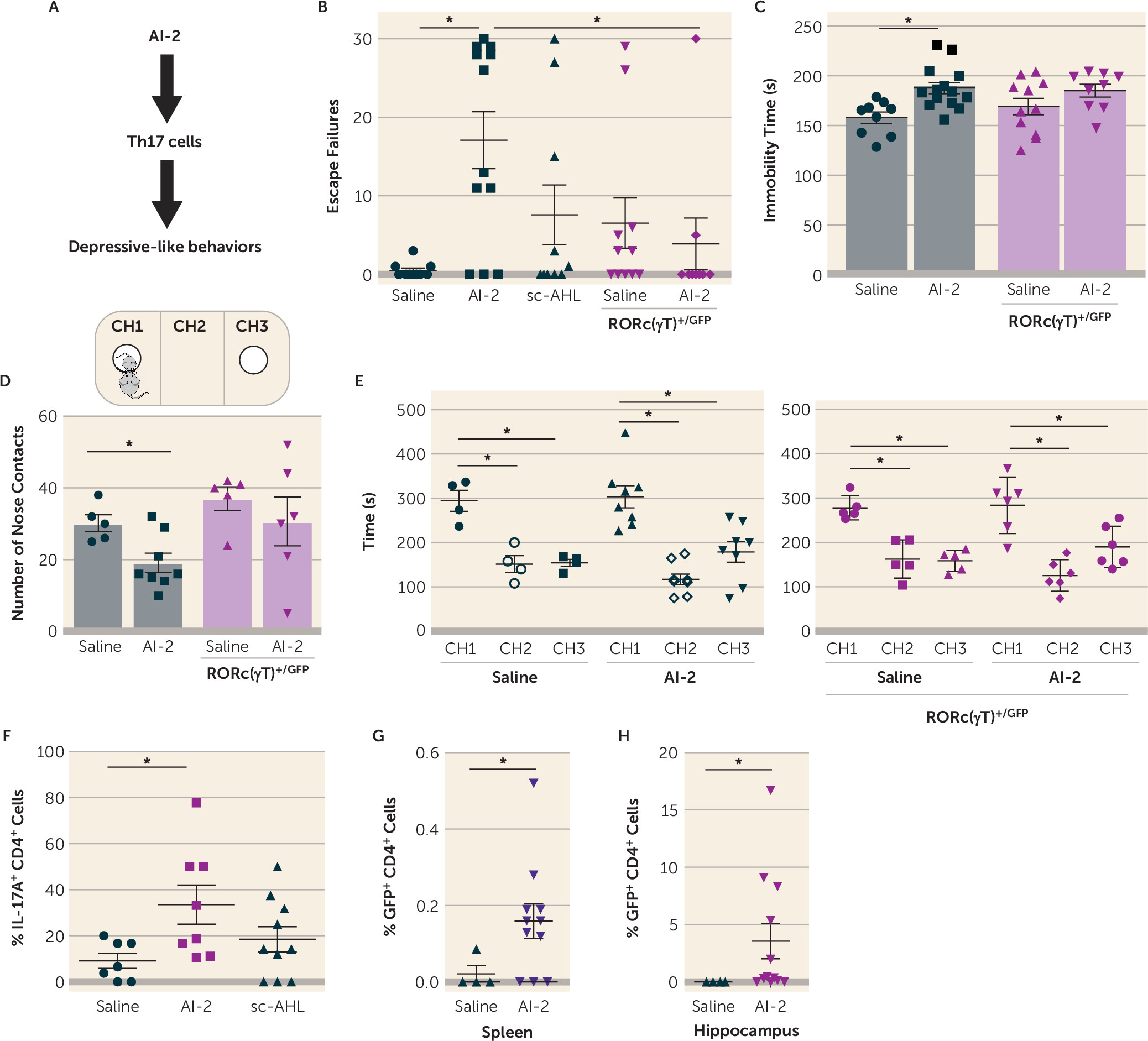
Since AI-2 was associated with increased susceptibility to learned helplessness in JAX mice, we tested whether AI-2 is sufficient to promote depressive-like behaviors (
Figure 3A) by daily treatments of AI-2 for 3 days before testing mice in the reduced paradigm of learned helplessness. With this paradigm, most mice do not develop learned helplessness (
40,
45). AI-2 administration, but not sc-AHL, was sufficient to increase susceptibility to learned helplessness in both TAC mice (
Figure 3B) and JAX mice (see Figure S1A in the
online supplement). Similarly, administration of AI-2 increased immobile time in the tail suspension test (
Figure 3C), often interpreted as an indication of increased despair or depression. In a measure of sociability, which is frequently impaired in depression and models of depression, administration of AI-2 reduced the number of nose contacts with an unfamiliar mouse (
Figure 3D), without interfering with the preference for the CH1 chamber containing the new mouse (
Figure 3E). AI-2 administration did not alter locomotor activity in a novel open field (see Figure S1C–E in the
online supplement). To determine whether bacteria were required for AI-2 prodepressive effects, we treated TAC mice and JAX mice with antibiotics to deplete the microbiota, and measured behaviors. Antibiotic treatment significantly depleted the microbiota, as shown by the reduced
Eubacterium levels (see Figure S1F in the
online supplement), and prevented AI-2 from promoting depressive-like behaviors (see Figure S1G–H in the
online supplement), suggesting that AI-2 acts on bacteria rather than the host to promote depressive-like behaviors. Taken together, these findings demonstrate that AI-2 administration increased displays of multiple depressive-like behaviors that are microbiome dependent.
SFB is well established as promoting production of Th17 cells (
34), which promote depressive-like behaviors (
40,
45), and Th17 cells accumulate in the hippocampus of mice exhibiting learned helplessness (
45) and are localized in the brain parenchyma (see Figure S2A in the
online supplement). Introduction of SFB in JAX mice increased the accumulation of hippocampal Th17 cells after learned helplessness (see Figure S2B–D in the
online supplement), whereas there was no difference in splenic Th17 cells (data not shown). We next investigated whether commensal-antigen-specific CD4
+ cells are sufficient to promote learned helplessness in SFB
+ Rag2
−/− mice. CD4
+ cells from mice expressing a transgenic T cell receptor specific for SFB-encoded antigen (7B8) or CD4
+ cells from littermate wild-type mice were adoptively transferred to Rag2
−/− mice and subjected to the reduced paradigm of learned helplessness. We found that 75% of Rag2
−/− mice receiving 7B8 CD4
+ cells exhibited learned helplessness, with an average of 23 escape failures, whereas none of the Rag2
−/− mice receiving wild-type CD4
+ cells exhibited learned helplessness, averaging 0.5 escape failures (see Figure S2E in the
online supplement). Consistently, hippocampal Th17 cells, but not Th1 cells, were significantly increased in Rag2
−/− mice receiving 7B8 CD4
+ cells and exhibiting learned helplessness, whereas receiving 7B8 CD4
+ cells without receiving foot shocks was not sufficient to increase hippocampal Th17 cells (see Figure S2F–G). Thus, the presence of SFB in the microbiota represents one factor that modulates Th17 cell trafficking during depressive-like behaviors.
Since SFB promotes learned helplessness, promotes hippocampal Th17 cell accumulation during learned helplessness, and produces AI-2, we tested whether AI-2 administration increases Th17 cells in the hippocampus. After administration of AI-2, mice were subjected to the reduced paradigm of learned helplessness because this paradigm does not increase hippocampal Th17 cell accumulation (
45), allowing for studies of factors that promote this outcome. Th17 cells were increased threefold in the hippocampus of mice receiving AI-2 compared with vehicle- or sc-AHL-treated mice (
Figure 3F), whereas AI-2 had no effect in the absence of foot shocks (see Figure S2H in the
online supplement) or in mice treated with antibiotics to deplete the microbiota (see Figure S2I in the
online supplement), demonstrating that AI-2 promotion of Th17 cell accumulation in the hippocampus is dependent on stress (foot shocks) and the microbiota. Furthermore, even though AI-2 did not promote Th17 cell differentiation in vitro (data not shown), AI-2 administration promoted Th17 cell differentiation in vivo, as indicated by the finding that AI-2 administration increased the percentage of GFP
+ CD4
+ cells both in the spleen (
Figure 3G) and the hippocampus (
Figure 3H) after learned helplessness in RORc(γT)
+/GFP mice, where a GFP reporter cDNA was knocked-in at the site of initiation of RORγt translation, leading to an important decrease in Th17 cells (
40,
44,
45), indicating that AI-2 was sufficient to induce Th17 cells after shocks. Since AI-2 promotes both Th17 cell differentiation and depressive-like behaviors, we tested whether Th17 cells mediate the depression-promoting effect of AI-2 by using RORc(γT)
+/GFP mice. RORc(γT)
+/GFP mice are resistant to the induction of learned helplessness, but adoptive transfer of Th17 cells is sufficient to resensitize RORc(γT)
+/GFP mice to learned helplessness (
41). The prodepressive effects of AI-2 administration were abolished in RORc(γT)
+/GFP mice (
Figure 3B–D), and this was independent of SFB, AI-2, sc-AHL, lc-AHL, or SAA levels (see Figure S3 in the
online supplement) or locomotor activity (see Figure S1C), which were similar in untreated wild-type and RORc(γT)
+/GFP mice. This indicates that Th17 cells are required to mediate AI-2-induced depressive-like behaviors.
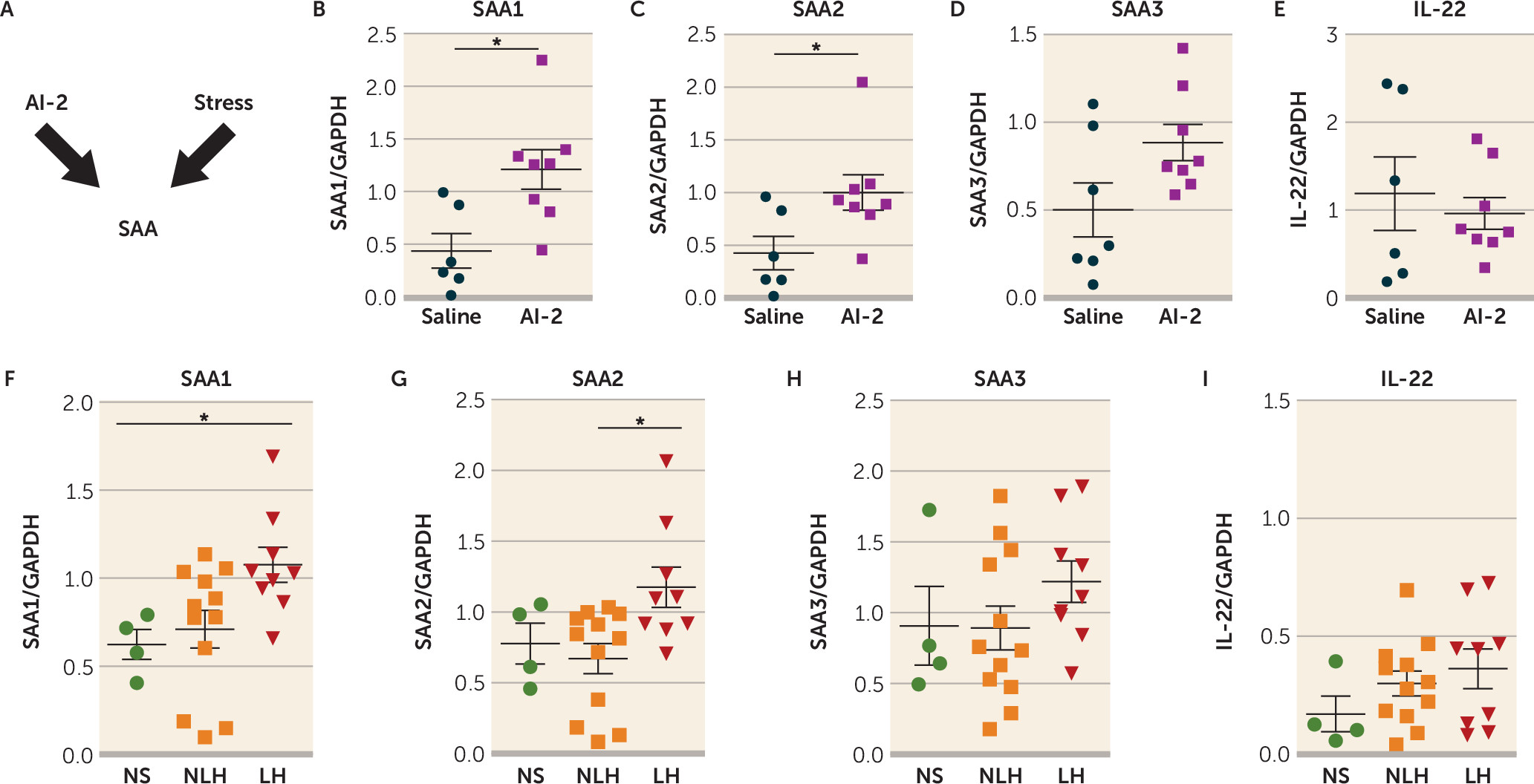
Because Th17 cells primarily reside in the lamina propria of the small intestine in healthy mice (
34), and SFB promotes Th17 cell differentiation in the small intestine via the SAA1-2/IL-22 pathway (
34,
35,
55), we tested whether intestinal SAA1, SAA2, SAA3, and IL-22 levels were affected by administration of AI-2 in TAC mice after stress (
Figure 4A). We found that AI-2 administration increased the intestinal expression of SAA1 and SAA2 (
Figure 4B–C) but did not change the intestinal expression of SAA3 and IL-22 (
Figure 4D–E) and that antibiotic treatment abolished this effect (see Figure S4A–B in the
online supplement). Furthermore, mice exhibiting prolonged learned helplessness (4 weeks) also exhibited higher intestinal levels of SAA1 and SAA2 (
Figure 4F–G), but not SAA3 and IL-22 (
Figure 4H–I), compared with mice that did not exhibit learned helplessness or non-shocked mice. Consistent with the report of Ivanov et al. (
34), SFB administration increased intestinal SAA1, SAA2, SAA3, and IL-22 levels (see Figure S4C–F). Furthermore, there were reductions of SAA1 and SAA2 levels after induction of learned helplessness in RORc(γT)
+/GFP mice compared with wild-type mice (see Figure S5A–D in the
online supplement), suggesting that RORc(γT)
+/GFP mice that are resilient to learned helplessness also exhibit lower levels of SAA1 and SAA2. Furthermore, injection of recombinant SAA2 increased susceptibility to learned helplessness by 50% (see Figure S5E). Taken together, these findings suggest that the increase of intestinal SAA1 or SAA2 levels after AI-2 administration might be sufficient to increase susceptibility to learned helplessness.
Since SAA promotes intestinal cytokine production, serum cytokine and chemokine levels were measured 3 to 48 hours after AI-2 treatment to determine whether inflammation might be contributing to the induction of Th17 cells and depressive-like behaviors after AI-2 treatment. The prototypical proinflammatory cytokines known to induce depressive-like behavior—IL-1, IL-6, and TNF (
36,
38)—were not affected by AI-2 treatment (see Figure S6 in the
online supplement). However, there were increased serum levels of IL-2, IL-13, IL-17A, and G-CSF and decreased levels of IL-4, IL-5, IL-12, GM-CSF, CXCL1, CCL2, and CCL5 after AI-2 treatment (see Figure S6) suggesting a remodeling of the inflammatory landscape by AI-2.
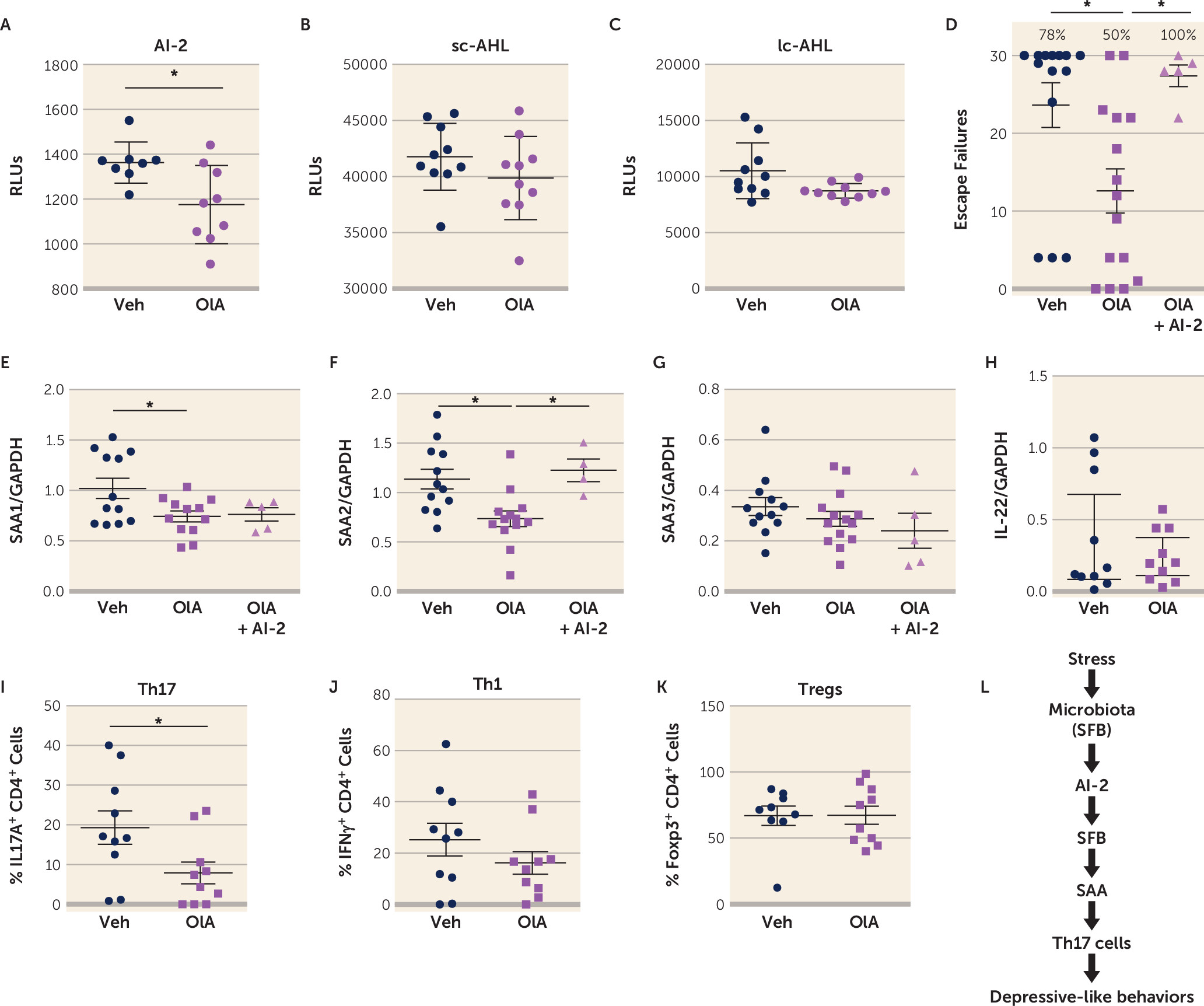
A screen of natural compounds identified oleic acid as a potential AI-2 inhibitor (
56). We tested whether in vivo administration of oleic acid prevented the production of AI-2 and the induction of learned helplessness. Oleic acid administration reduced fecal levels of AI-2, but not sc-AHL or lc-AHL in mice subjected to the learned helplessness paradigm (
Figure 5A-C), confirming the ability of oleic acid to inhibit AI-2 production in vivo. Oleic acid reduced the susceptibility to learned helplessness of TAC mice from an average of 24 failures out of 30 trials to 13 failures, and it reduced the percentage of learned helpless mice from 78% to 40%, suggesting an antidepressant action of oleic acid (
Figure 5D). The coadministration of AI-2 with oleic acid abolished this antidepressant effect, resulting in an average of 27 failures out of 30 trials (
Figure 5D), demonstrating the requirement of AI-2 in the antidepressant effect of oleic acid. As expected, levels of factors downstream of AI-2, SAA1, and SAA2, but not SAA3 or IL-22 levels, in the intestine were reduced by oleic acid administration (
Figure 5E–H) after learned helplessness. However, only SAA2 levels were restored to vehicle levels after AI-2 coadministration, suggesting that SAA2 mediates AI-2-dependent depressive-like behaviors. Furthermore, the percentage of Th17 cells was also reduced in oleic acid–treated mice, whereas Th1 and regulatory T cells (Tregs) were not affected (
Figure 5I–K). This reduction of Th17 cells was not associated with changes in gut permeability (see Figure S7 in the
online supplement). Altogether, these data provide evidence for a new circuit involving SFB/AI-2/SAA2/Th17 cells in promoting depressive-like behaviors after stress in mice (
Figure 5L).
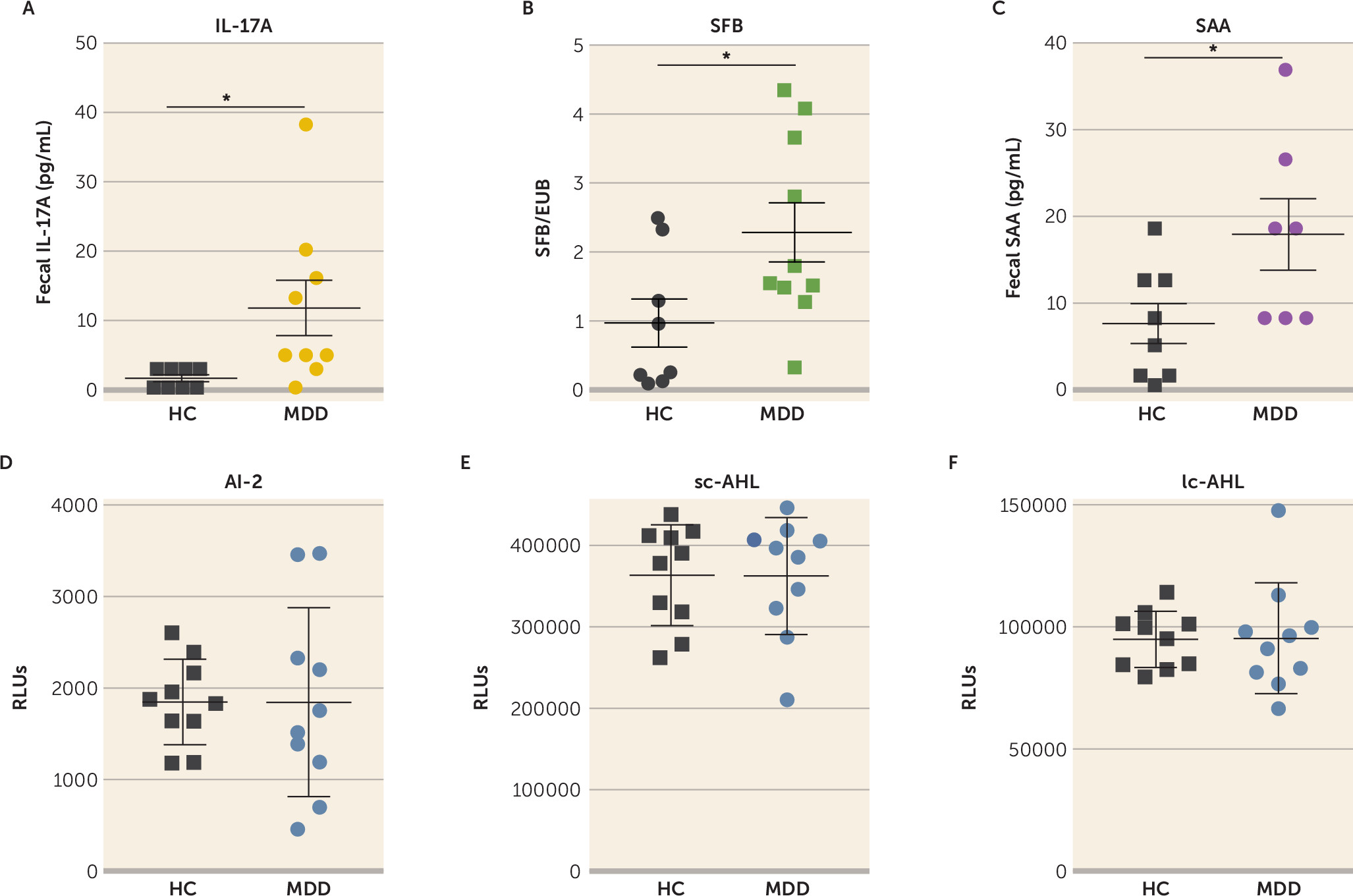
To determine whether the findings in mice are relevant for humans, we analyzed the levels of IL-17A, SAA, AI-2, and SFB in the stools of 10 patients with a current primary diagnosis of major depressive disorder and 10 healthy matched subjects. We found that the IL-17A level was increased approximately eightfold in patients with current major depression compared with healthy subjects (
Figure 6A), and SAA and SFB levels were increased approximately twofold (
Figure 6B, C), whereas AI-2, sc-AHL, and lc-AHL levels did not differ between groups (
Figure 6D–F). Furthermore, regression analyses showed that AI-2 levels were predicted by the presence of fecal SFB in patients with current major depression and healthy subjects; that is, SFB levels predicted AI-2 levels (r=0.494, p=0.019) (
Table 1). Similarly, SFB levels predicted SAA levels (r=0.336, p=0.004), SAA levels predicted IL-17A levels (r=0.288, p=0.011), and IL-17A levels predicted QIDS scores (r=0.245, p=0.013) (
Table 1). No interaction of SFB with sc-AHL or lc-AHL was observed (p=0.335 and p=0.217, respectively). These data suggest that patients with current major depression have a dysregulation of the intestinal SFB/AI-2/SAA/IL-17A circuit.
Discussion
We uncovered a novel mechanistic circuit linking the gut microbiome and bacterial products to the brain axis that regulates depressive-like behaviors in mice. SFB in the gut increases the bacterial QSM AI-2 and the levels of SAA1 and SAA2, and these increase the production of Th17 cells that promote depressive-like behaviors (
55) (
Figures 2–
4). Furthermore, administration of either SFB or AI-2 is sufficient to increase susceptibility to depressive-like behaviors of SPF mice with an intact microbiome. We also found that disrupting this signaling pathway by blocking AI-2 with oleic acid provides antidepressant properties. These findings reveal a signaling mechanism by which changes in bacterial products influence mood-relevant behavior.
As has previously been reported, microbiota composition was found to influence behaviors (
2,
8,
57). We identified SFB, a spore-forming bacteria belonging to the Firmicutes phylum and closely related to the Clostridium family, as candidate bacteria that influence mood-relevant behaviors. This extends previously published reports that the Firmicutes are modulated in mice after stress exposure (
9,
25,
58). Shifts in the composition of the two predominant phyla of the gut microbiome, Bacteriodetes and Firmicutes, have been associated with several CNS conditions (e.g., autism, stress, and Alzheimer’s disease) (
59,
60). Although the composition of the gut microbiota has been associated with depressive disorders, the mechanisms whereby gut bacteria modulate mood remain largely unknown (
25,
61). We propose that induction of Th17 cells by SFB in the gut increases susceptibility to depressive-like behaviors. However, it is not excluded that other bacteria (such as
Akkermansia muciniphila and
Mucispirillum schaedleri, the latter being shown in another study [
62] to be decreased after stress) are also involved in the modulation of depressive-like behaviors.
Akkermansia muciniphila is known for its anti-inflammatory properties (
63), which could also modulate the inflammation associated with depressive-like behaviors (
36,
38). In addition to bacterial abundance, interactions between bacteria may be important in regulating behaviors, although further experiments are needed to test this hypothesis.
Our evidence indicates that the bacterial quorum-sensing molecule AI-2 can be an important conduit of signals from the gut to the brain. This was particularly evident from the finding that AI-2 administration was sufficient to promote depressive-like behaviors, which also demonstrated that gut bacterial metabolites have the capacity to modulate behaviors in SPF mice. In addition, increased availability of AI-2 increases the ratio of Firmicutes to Bacteriodetes, favoring the Firmicutes (
64), which may create a vicious circle, amplifying depressive-like behavior. AI-2 is produced by SFB bacteria and is a key link between SFB and the production of Th17 cells, one mechanism by which SFB and AI-2 can promote susceptibility to depressive-like behaviors. However, we found that AI-2 did not act directly on the host, demonstrating that AI-2 mediates its effects through bacteria. Levels of AI-2 (
Figure 2) but not SFB (data not shown) increased after foot shocks, suggesting that either foot shocks specifically affect SFB-dependent production of AI-2 or, more likely, other bacteria producing AI-2 are affected by foot shocks and are responsible for the increased production of AI-2. We also found increased SFB levels in AI-2-treated JAX mice after foot shocks, suggesting that AI-2 is able to increase the population of SFB (see Figure S1B in the
online supplement), which contributes to the increased AI-2 production. However, because other bacteria also produce AI-2, this signaling circuit is not limited to SFB (
65). Although AI-2 is normally produced in the lumen of the intestines, we used intraperitoneal administration, so further studies using intraluminal administration would more closely model its endogenous mode of production.
Reports have shown the presence of AI-2 and AHLs in humans, in particular in patients with inflammatory bowel disease (
65,
66), who are known to have an elevated risk of comorbid depression (
67). Consistent with this, we were also able to detect AI-2 in the stools of patients with current major depression. To our knowledge, quorum-sensing molecules have not previously been shown to modulate behaviors, but they are critical in regulating biofilm formation and bacterial virulence (
53,
54). Therefore, the identification of AI-2 as a modulator of depressive-like behaviors represents a novel discovery that may help clarify mechanisms by which the gut microbiome influences behavior and mood.
Widely distributed and abundant in nature, oleic acid is a monounsaturated omega-9 fatty acid that was reported to be deficient in patients with major depression (
68) and increased in stress-resilient rats (
69), suggesting that oleic acid helps in coping with stress. Oleic acid has also been associated with the maintenance of mental well-being (for reviews, see references
70–
72) and treatment response to the antidepressant imipramine (
73). Intake of oleic acid in women was associated with lower risk of severe depressed mood (
74). Oleic acid is the predominant monounsaturated fatty acid in olive oil, and it has been proposed to contribute to the beneficial effect of olive oil (
75). We confirmed that oleic acid is an AI-2 inhibitor (
56) and that oleic acid exhibited antidepressant properties in the learned helplessness model of depression, which could be reversed by the addition of AI-2. This showed that the antidepressant effects of oleic acid were indeed AI-2 dependent.
Interest in the role of Th17 cells in depression has been increasing during the past several years (
76). Th17 cells are usually thought of as being pathogenic in autoimmune diseases (
76). Th17 cells are predominantly localized in the lamina propria of the small intestine in healthy mice, resulting from the activation of an IL-22-ILC3-SAA1/2 pathway induced in response to SFB, which only colonizes the ileum (
55). We found that SAA1 and SAA2, but not SAA3, increase in mice exhibiting learned helplessness and in mice receiving AI-2, and decrease after oleic acid treatment, suggesting that the SAA1/2 pathway is induced after AI-2 treatment. Consistent with this, we found that SAA2 is sufficient to increase susceptibility to learned helplessness, and reintroduction of AI-2 in oleic acid–treated mice increased SAA2 but not SAA1. However, we did not find any difference in IL-22 levels after learned helplessness or AI-2 treatment, and IL-22 neutralization did not provide any antidepressant action (data not shown), suggesting that the ILC3 cells may not be involved in promoting Th17 cell production after stress. Altogether this suggests that after stress, AI-2 promotes the differentiation of Th17 cells independently of the IL-22-ILC3 pathway and that the IL-22-ILC3 pathway may be necessary only during the step of colonization by SFB to induce Th17 cells (
55). Nevertheless, AI-2 promotes SAA1 and SAA2 production, leading to increased Th17 cell production (
55) and increased hippocampal Th17 cell accumulation. Even though Th17 cells accumulate in the hippocampus after AI-2 treatment, it remains to be determined whether the Th17 cells produced in the intestine are indeed migrating to the brain. We found that AI-2 promotes Th17 cells in the brain, oleic acid reduces hippocampal Th17 cells, and SFB-specific T cell receptor CD4
+ cells promote depressive-like behavior in Rag2
−/− mice, increasing hippocampal levels of Th17 cells, supporting the idea that intestinal Th17 cells migrate to the hippocampus. However, further experiments are required to determine the role of hippocampal Th17 cells in promoting depressive-like behaviors, the migratory mechanisms whereby Th17 cells exit the intestine after stress, and how intestinal Th17 cells infiltrate the brain. Indeed, we did not find differences in gut permeability after AI-2 or oleic acid treatments compared with controls, whereas both AI-2 and oleic acid modulate the levels of hippocampal Th17 cells and behaviors. Therefore, it is possible that rather than changing gut permeability, and because AI-2 could not be detected in the blood (data not shown), AI-2 induces changes in Th17 cell migratory properties, such as the production of chemokine factors that attract Th17 cells outside of the gut. Consistent with this idea, we found changes in serum chemokine levels after AI-2 administration, but further experiments will be needed to answer this question. It is important to note that Th17 cells were recently shown to promote maternal immune activation–associated autistic traits by inducing IL-17A-dependent neurodevelopmental abnormalities in the offspring (
77), suggesting that the cytokine IL-17A is sufficient to mediate some of the effects of Th17 cells in autism. The induction of Th17 cells was dependent on the presence of SFB in the ileum of the pregnant mothers (
47), suggesting an important SFB/Th17 circuit in priming behaviors, even though the outcome behaviors may differ depending on the timing of induction and the location of Th17 cells.
Th17 cells have been shown to be induced by a high-salt diet (
78) and to mediate dietary salt–induced neurovascular and cognitive impairments (
79), reinforcing the idea that healthy diet, by preventing the increase of Th17 cells, enhances mental health.
Furthermore, we found that fecal IL-17A levels were increased in patients with current major depression and predicted QIDS scores, reinforcing the previously found contribution of Th17 cells to major depressive disorder (
80,
81). Associated with a potential intestinal Th17 cell increase in major depression were increased fecal levels of SFB and SAA, whereas QSM levels did not change. This suggests that targeting the Th17 cell pathway may be sufficient to improve major depression. However, our study’s sample size was small, and larger investigations are required.
Together, these data uncovered a novel SFB/AI-2/SAA1-2/Th17 cell pathway that promotes depressive-like behavior, and reducing AI-2 may provide therapeutic benefit.