Depression is a highly prevalent health condition that incurs great personal, social, and economic burdens, making it a leading cause of disability worldwide (
1). Thus, developing effective strategies for prevention of depression could considerably reduce the global burden of mental illness (
2). This concept has inspired a vast amount of research aiming to identify modifiable risk factors for depression. Unfortunately, targeting modifiable risk factors for mental illness has so far proved limited and presents several difficulties. For instance, a 2018 “umbrella review” covered a plethora of lifetime risk factors for depression, including 134 meta-analytic syntheses from 1,283 primary studies (
3). Among this mass of literature, however, only a small handful of factors had convincing prospective evidence, related to things such as widowhood, high risk of metabolic diseases, childhood abuse, sexual dysfunction, and job strain. Furthermore, such evidence is also unable to determine whether said risk factors are
causally related to depression, for several reasons, such as reverse causation and residual confounding (
4–
6). Of course, the gold-standard approach to determining causality is the randomized controlled trial. However, conducting a well-powered randomized controlled trial to examine the casual impact of certain risk factors can be unethical (for instance, with smoking) or highly time consuming and expensive, especially in the absence of any previous indication of causal associations (
4,
6).
In this issue of the
Journal, Choi and colleagues (
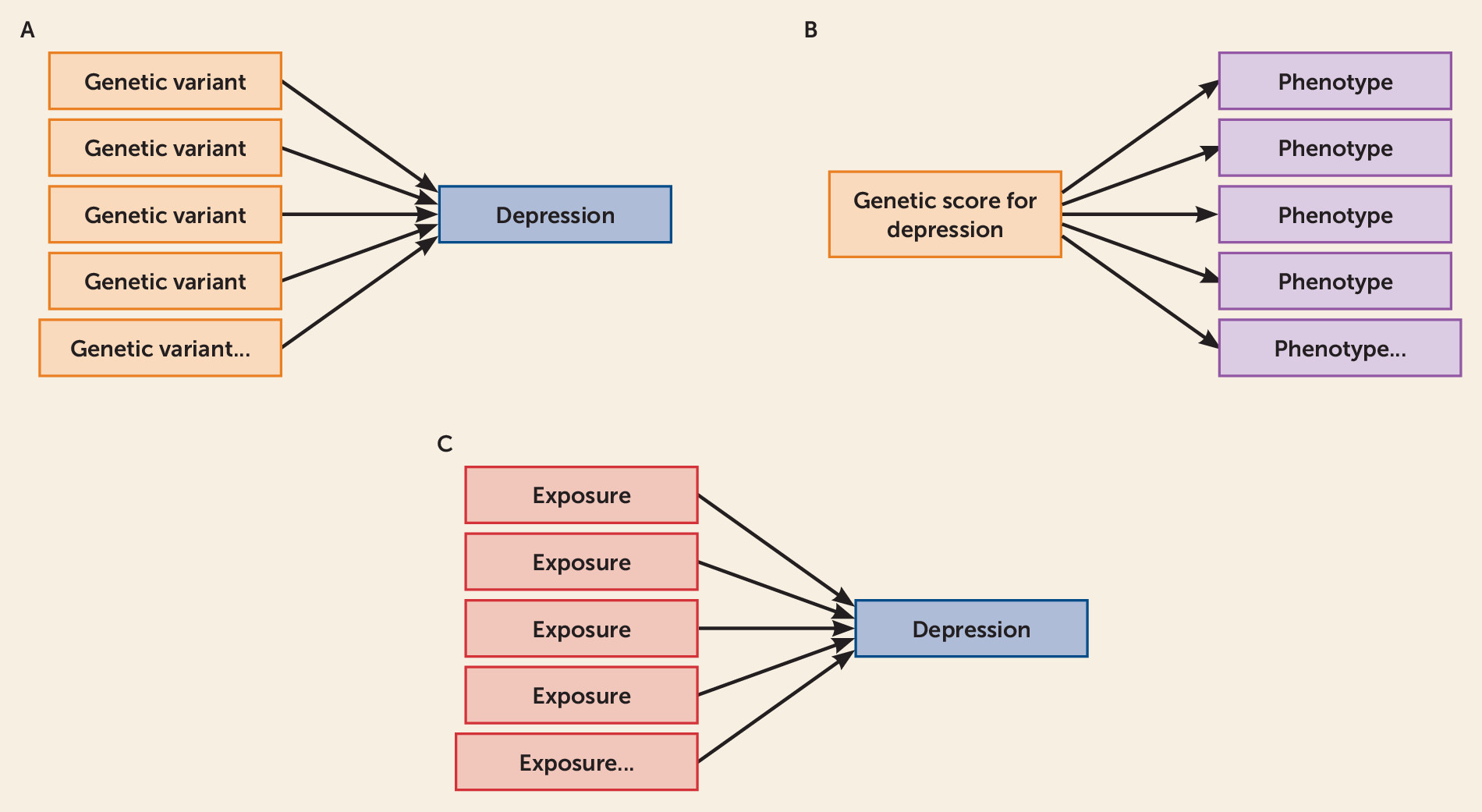
5) set out a novel approach to identifying causal factors for depression. They do this through capitalizing on the rich data of the UK Biobank, along with new statistical methods using a powerful, two-stage approach to triangulate their findings and strengthen causal inferences. In stage 1, they employ a relatively hypothesis-free approach to identifying multiple possible risk and protective factors for depression from over 100 UK Biobank variables. The authors refer to this as an “exposure-wide association scan,” which is conducted in the style of a phenome-wide association study and a genome-wide association study, only instead of identifying associated phenotypes or associated genetic variants, the exposure-wide association scan specifically identifies modifiable exposures associated with the outcome of interest—in this case depression. A comparison of these hypothesis-free methods is provided in
Figure 1.
Of the 106 modifiable risk factors explored in Choi and colleagues’ exposure-wide association scan, 29 were associated with increased risk of depression in the fully adjusted model, mostly related to social engagement, physical activity, media use, and diet. The broad initial approach employed within the exposure-wide association scan proved beneficial in several ways, allowing Choi et al. to identify “new” factors given little or no attention previously (e.g., tea drinking), while confirming associations with previously studied risk factors (e.g., social support) in a hypothesis-free context, and also calling into question some previously investigated factors that did not associate in this context.
In stage 2, the modifiable factors indicated from stage 1 were followed up using summary-level “Mendelian randomization” analyses. Mendelian randomization examines potential causal effects between a modifiable exposure and an outcome. It is an extension of instrumental variable analysis. The instrument takes the form of genetic variants that serve as proxies for the exposure (i.e., the modifiable protective and risk factors examined) (
6). If the genetic variants predict the outcome (in this case, depression), this could indicate a causal effect. However, three crucial assumptions must be satisfied: 1) the genetic variants are robustly associated with the exposure, 2) the genetic variants are not associated with confounders of the exposure-outcome relationship, and 3) the genetic variants only influence the outcome through the exposure. Since genetic variants are “randomized” at conception, and they alter average lifetime levels of the exposure, examining their impact on the outcome in this way is akin to a natural experiment (
6). Our genes also remain unchanged throughout our lifetime, meaning that they cannot be altered by the outcome of interest, hence reducing bias from reverse causation. Any causal effects suggested using Mendelian randomization could then be followed up using randomized controlled trials and intervention trials.
Of the 29 associated risk factors identified in the stage 1 exposure-wide association scan, the stage 2 Mendelian randomization provided evidence of possible causal effects for four: multivitamin intake, daytime napping, and TV use increased depression risk, and higher frequency of “confiding in others” exerted a protective effect. However, a particularly important limitation of Mendelian randomization in psychiatric research is horizontal pleiotropy (
7), which occurs when one genetic variant has multiple effects on several possible phenotypes. This can change levels of the outcome through pathways other than via the exposure, thus violating the core assumptions of Mendelian randomization and reintroducing confounding. Because psychiatric phenotypes are highly polygenic and the biological function of specific genetic variants is largely unknown, it is not usually possible to rule out pleiotropic pathways—so instead Choi et al. applied sensitivity analyses more robust to pleiotropy (specifically, Mendelian randomization-Egger [MR-Egger] and weighted median [
8,
9]) to assess how this influenced their findings. Whereas these analyses called into question the associations between depression and multivitamin usage (suggesting that these results may be due to pleiotropic pathways), the indicated casual effects of daytime napping, TV use, and “confiding in others” on depression were consistent across all pleiotropy-robust sensitivity analyses.
With regard to daytime napping, Choi and colleagues’ analyses also found strong evidence for depression liability increasing daytime napping, suggesting that the two present a vicious circle. While this could be interpreted in the context of the known bidirectional relations between sleep (i.e., as a broader construct) and mental health outcomes (
10), Mendelian randomization analysis does not speak to the pathways underlying the relations between a specific exposure like daytime napping and the target outcome (only to causality and directionality). Furthermore, actual sleep duration did not show any evidence of a causal effect in the Mendelian randomization analysis, although such analyses only look for linear effects, and therefore this must also be considered alongside evidence that associations between sleep duration and mental illness can be nonlinear (
10,
11).
While higher levels of TV use were also found to increase depression risk, in this case there was no evidence for reverse causation. These causal effects could possibly be underpinned by TV use acting as a proxy for sedentary behavior, as there is emerging observational and experimental evidence implicating high levels of sedentariness in onset of depressive symptoms (
11,
12). However, no significant causal effects on depression were found for computer use (another sedentary behavior), although individuals with higher depression risk were more likely to spend time using computers. Such conflicting findings should be considered alongside the nature of the brief self-report measures used to capture these and many of the variables examined in the Choi et al. study, which remains a major limitation to how the findings can be interpreted, given the strong possibility that such self-report data may not accurately reflect individuals’ actual lifestyles and environments. For instance, in relation to physical activity and sedentary behaviors, previous Mendelian randomization studies have indicated that objective measures (such as accelerometry) may ultimately be required to accurately capture causal relations with mental health (
13).
Nonetheless, compelling evidence for “frequency of confiding in others” as a protective factor for depression was found in the whole sample, in high risk groups, and in the Mendelian randomization analysis. It was also consistent across the Mendelian randomization sensitivity methods, with no evidence of pleiotropy or reverse causation. This finding demonstrates robust triangulation suggesting that interventions that provide social support (and promote the development of meaningful social relationships) are an important target for preventing depression in both the general population and those at increased risk.
However, one consideration is that the approaches applied here might not be powerful enough to detect some of the small but meaningful effects of risk factors that do not survive correction for multiple testing. Therefore, with regard to the “null” variables found in this study, one must always bear in mind that absence of evidence is not evidence of absence. Furthermore, Mendelian randomization only looks at lifelong effects, whereas some causal factors in depression onset might exert more acute effects. Relatedly, it is important to remember that whereas the majority of mental health conditions first present during adolescence or early adulthood, many large-scale health resources with sufficient power for Mendelian randomization analyses (such as the UK Biobank) contain data from mostly adult/older adult populations.
While focusing here on the implications for depression prevention, we stress our enthusiasm about the possibility that the novel two-stage exposure-wide association scan/Mendelian randomization approach applied by Choi et al. could be used for other psychiatric outcomes in the future. Indeed, the hypothesis-free approach may suit psychiatric epidemiology particularly well, since mental health conditions may have much more diverse risk factors than physical health conditions (
2). As they are so intertwined with the environment, their risk factors tend to change and evolve, making hypothesis-free methods useful in capturing these diverse predictors, at both the individual and societal levels (
2). For effective interventions, large numbers of related risk factors may need to be considered, and Choi et al. suggest that hypothesis-free approaches can be highly valuable for identifying multiple risk factors that could work together in an intervention package. Although not done here, this method could be extended by using multivariable Mendelian randomization and factorial Mendelian randomization methods, which test mediation pathways and cumulation of risk factors, respectively (
7). Alongside this, further research in younger samples is needed to confirm the role of putative risk factors in primary prevention of depression. Nonetheless, robust Mendelian randomization studies such as that of Choi et al. are providing new and promising routes toward identification of causal risk factors for depression. Continued progress in this area is of paramount relevance for the development of targeted interventions to prevent depression and reduce the global burden of mental illness.
Acknowledgments
Dr. Firth is supported by a University of Manchester Presidential Fellowship (P123958) and a UK Research and Innovation Future Leaders Fellowship (MR/T021780/1) and has received support from an NICM-Blackmores Institute Fellowship. Dr. Wootton is funded by the Wellcome Trust (204895/Z/16/Z) and works in a unit that receives funding from the UK Medical Research Council (MC_UU_00011/1 and MC_UU_00011/3).