Can cellular and molecular drivers of posttraumatic stress disorder (PTSD) and depressive disorders be disambiguated? Roughly half of individuals with PTSD will develop major depressive disorder. In addition to their frequent comorbidity, both PTSD and major depressive disorder are more commonly diagnosed in women and have overlapping DSM-defined symptoms. They are heterogeneous both in origin and in the people they affect; for example, while each can be triggered by trauma, what constitutes a traumatic event varies, and trauma will trigger disease only in some individuals. Ultimately, the combination of comorbidity and heterogeneity within PTSD and major depressive disorder has hindered our understanding of each disorder’s genetic underpinnings.
In this issue of the
Journal, Jaffe et al. (
1) took a powerful Jaccardian (i.e., gene set overlap) approach to the shared etiologies of major depressive disorder and PTSD, exploiting comparative transcriptomics in human postmortem brain tissue to discover molecular similarities and differences. Their gene and gene network analyses focused on previously implicated brain regions within the cortico-limbic circuit, including the dorsal anterior cingulate cortex, dorsolateral prefrontal cortex, basolateral amygdala, and medial amygdala. This hypothesis-free transcriptomic view led to further insights into how each region may be involved, as well as to several unexpected findings and important takeaways for these frequently comorbid disorders.
Surprisingly, in individuals with PTSD compared to neurotypical control subjects, transcriptome differences attributable to PTSD were not subregion driven, and more differentially expressed genes (DEGs) were identified in cortex, despite the amygdala’s role in mediating fear and negative emotionality.
CRHBP (corticotropin releasing hormone binding protein) was the only amygdala DEG, while
HDAC4 (histone deacetylase 4),
SPRED1 (Ras/MAPK signaling pathway gene), and
CORT (cortistatin) were among the top 41 DEGs identified in the cortex. At a slightly relaxed DEG significance,
CORT was also downregulated in both the basolateral amygdala and medial amygdala subregions. Despite DEG differences between cortex and amygdala, gene ontology findings largely functionally converged, implicating immune-related gene network disturbances in PTSD. The authors then used single-nucleus RNA sequencing and RNAscope in brain tissue from neurotypical control subjects to localize DEGs to the microglial and inhibitory neuron populations, ultimately observing high correlations between
CORT expression and somatostatin-positive neurons and
CRHBP coexpression with both
CORT and
GAD2 across brain regions. Taken together, these findings add to evidence that PTSD symptoms throughout the lifetime may be linked to chronic desensitization of the stress, immune, and GABAergic systems and support previous studies linking
CHRBP as well as
FKBP5 (
2) (although
FKBP5 was not implicated in the Jaffe et al. study) to PTSD, childhood trauma, and suicidality (
3).
In contrast to PTSD, the major depressive disorder transcriptome had substantially more DEGs in both cortex and amygdala, with the anterior cingulate cortex and medial amygdala subregions driving differential expression. These region-specific signatures of altered gene expression can help generate insights into the genesis and maintenance of major depressive disorder, and, potentially, novel targets for therapeutic intervention. Broadly, however, many of the major depressive disorder DEGs overlapped with PTSD (p<1×10−46), and immune-specific gene sets were highly concordant. Overall, major depressive disorder and PTSD transcriptomes seem to point to shared alterations in the stress, inhibitory signaling, and immune domains.
Given the potential for confounding factors, the functional congruences that Jaffe et al. observed between PTSD and major depressive disorder are remarkable. They speak to the existence of shared mechanisms that may, in part, lead to their high comorbidity. Nevertheless, the molecular changes observed are also representative of ongoing pathological processes underlying both disorders throughout the lifetime. While postmortem brain transcriptomics likely captures some of the active etiologic components, it cannot inform directly as to genetic causes or other critical events that may be observable only during development, at the time these diseases are triggered, or early in disease course.
This study illustrates both the opportunities and challenges of human postmortem brain transcriptomic studies for psychiatric disorders. On the one hand, the increasing accessibility and affordability of transcriptomic technologies now allows for broad transcript comparisons over time, between brain regions, and across patient populations, down to the resolution of a single cell. In the context of a well-controlled study, these molecular changes are observed in isolation, but they exist within the brain and body connectome, where even modest changes in single genes can drive behavior (
4). On the other hand, transcriptomic studies are limited by many factors, including the clinical covariates and heterogeneity of the brains representing any given diagnosis (
5), the technical and bioinformatic challenges of generating and analyzing sequencing data (
2), and the potential compensatory responses that these postmortem cellular snapshots may represent. Any of these factors could potentially speak to one of the more paradoxical findings in this study, namely, the
downregulation of several critical inflammatory genes and pathways in PTSD and major depressive disorder, counter to several observations that inflammatory processes are pathogenic in depression (
6). Although Jaffe et al. adjust for important population covariates such as sex, ethnicity, and substance use, a statistical correction does not necessarily equate to biological rectification (
3,
5). In fact, the overrepresentation of females with PTSD compared to female neurotypical control subjects brings this study’s findings in alignment with another recent study that observed a sex difference in inflammatory gene transcripts in female versus male PTSD patients (
5).
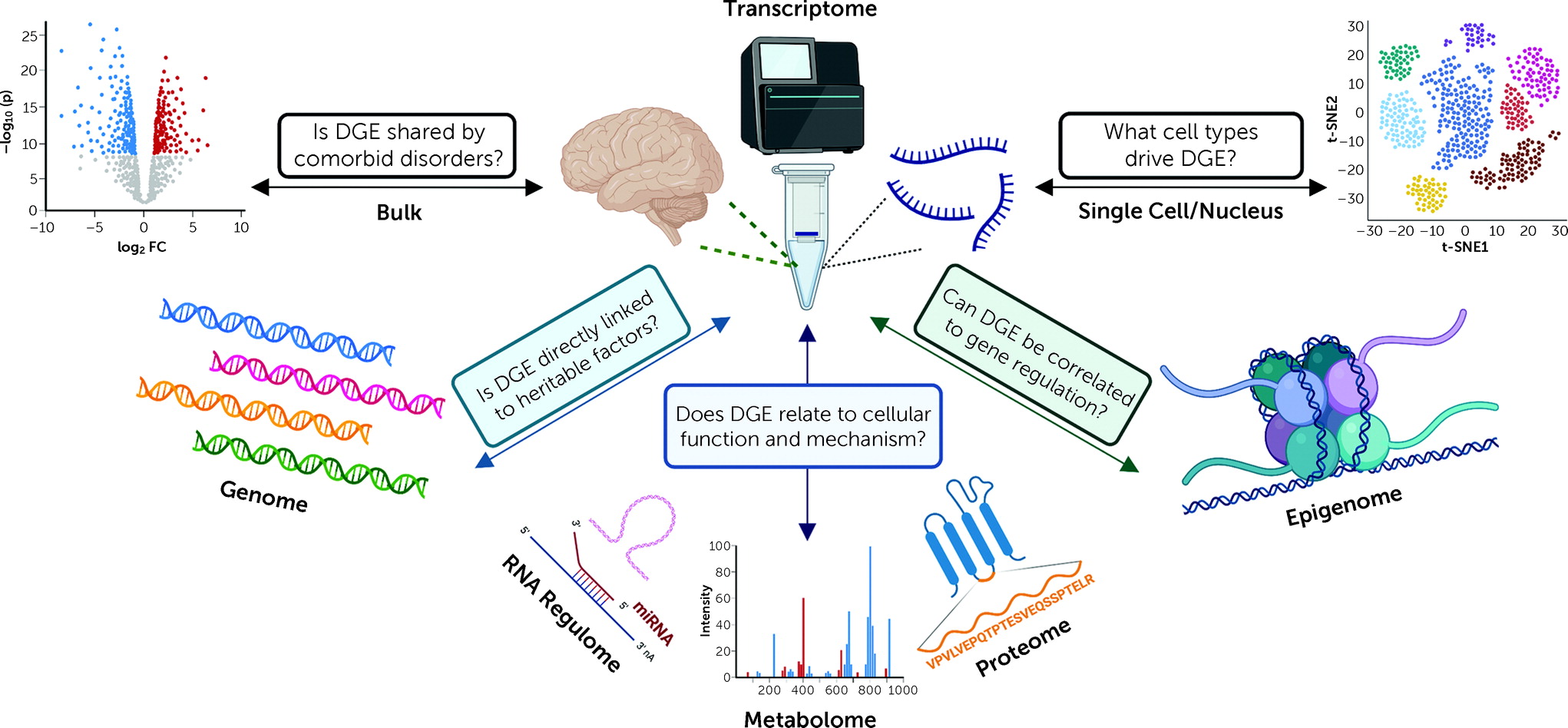
Each new study contributing transcriptomes to the information commons represents an invaluable contribution because of ways these data can be interconnected to other omic data sets (genome, metabolome, proteome, methylome, etc.) (
Figure 1). Resources such as the NeuroBioBank, a consortium of brain banks, enable multiple investigators to generate different molecular views of the same disorders, brain regions, and cells, within the same human postmortem brains. Together, these omic studies represent a bridge between human clinical studies and animal models, especially for psychiatric disorders, where animal-model counterparts have not yet been identified or have been questioned (
7). As more types of data sets are generated, longitudinal and “meta” comparative studies become possible, extending our understanding of the relationships of genes and gene networks to their causes and consequences in specific disorders, and their pleiotropy across disorders. In this way, identifying convergence between transcriptomic disease signatures parallels discoveries of shared inheritance, as was previously achieved via twin studies, and as is now increasingly done via polygenic scores from genome-wide association studies (
8). Moreover, divergences between transcriptomic signatures may help resolve the biological nuances of disorders that are phenotypically similar but have distinct onsets and triggers. For example, delineating differences in the molecular etiology of perimenopausal depression (
9) and other ovarian steroid–related mood disorders from those of major depressive disorder more generally could improve the specificity and efficacy of treatment (
10).
As might be expected for a technology that attempts to measure thousands of dynamically changing mRNA transcripts, results can be clouded by a variety of biological and technical factors. In addition to seeking predictive validity in the cellular and animal model studies that follow, it is vital to compare similar transcriptomic studies to each other to evaluate generalizability, replicability, and reliability. For major depressive disorder, interstudy comparisons are so far limited, but encouraging. Jaffe et al. compare their PTSD findings to a similar, albeit smaller, human postmortem brain study by Girgenti et al. (
2). Notably, their data largely support the findings of that earlier study, with a couple of key differences that are well detailed in the article. But underscoring the importance of expanding the quantity and diversity of postmortem brains available, many donors were shared across these two studies. Even though each examined different brain hemispheres and performed independent procedures, interpreting convergences in results critically relies on recognizing overlaps between study populations. More generally, in comparative and multi-omic studies, caution is warranted when using publicly accessible data resources, as the sources of the data may be related, or even identical. For some omic data, overlapping samples may be more easily discerned (e.g., genomics), but for others (e.g., lipidomics), duplicate samples are not yet routinely identified without other information.
While transcriptomics alone cannot answer all the questions surrounding comorbidity of PTSD and major depressive disorder, the characterization of these disease signatures, and the relation of their differences to other omic and functional measures, is foundational. Jaffe and colleagues’ study thus significantly expands our understanding of cortex and amygdala dysregulation in PTSD and major depressive disorder, having identified intriguing commonalities and differences in stress and immune gene networks that may speak to genetic drivers of these diseases. In the future, new insights into the origins of psychiatric disease are likely to emerge as these transcriptomic data are integrated into the greater molecular interactome.