Characterizing the Shared Genetic Underpinnings of Schizophrenia and Cardiovascular Disease Risk Factors
Publication: American Journal of Psychiatry
Abstract
Objective:
Schizophrenia is associated with increased risk of cardiovascular disease (CVD), although there is variation in risk among individuals. There are indications of shared genetic etiology between schizophrenia and CVD, but the nature of the overlap remains unclear. The aim of this study was to fill this gap in knowledge.
Methods:
Overlapping genetic architectures between schizophrenia and CVD risk factors were assessed by analyzing recent genome-wide association study (GWAS) results. The bivariate causal mixture model (MiXeR) was applied to estimate the number of shared variants and the conjunctional false discovery rate (conjFDR) approach was used to pinpoint specific shared loci.
Results:
Extensive genetic overlap was found between schizophrenia and CVD risk factors, particularly smoking initiation (N=8.6K variants) and body mass index (BMI) (N=8.1K variants). Several specific shared loci were detected between schizophrenia and BMI (N=304), waist-to-hip ratio (N=193), smoking initiation (N=293), systolic (N=294) and diastolic (N=259) blood pressure, type 2 diabetes (N=147), lipids (N=471), and coronary artery disease (N=35). The schizophrenia risk loci shared with smoking initiation had mainly concordant effect directions, and the risk loci shared with BMI had mainly opposite effect directions. The overlapping loci with lipids, blood pressure, waist-to-hip ratio, type 2 diabetes, and coronary artery disease had mixed effect directions. Functional analyses implicated mapped genes that are expressed in brain tissue and immune cells.
Conclusions:
These findings indicate a genetic propensity to smoking and a reduced genetic risk of obesity among individuals with schizophrenia. The bidirectional effects of the shared loci with the other CVD risk factors may imply differences in genetic liability to CVD across schizophrenia subgroups, possibly underlying the variation in CVD comorbidity.
Schizophrenia is associated with a two- to threefold greater risk of cardiovascular disease (CVD) compared with risk in the general population, contributing to a 10- to 20-year reduced life expectancy (1, 2). Despite increasing awareness of this comorbidity over recent decades, the level of CVD risk does not appear to have decreased for individuals with schizophrenia (3), and there has been little or no progress in decreasing the mortality gap (2, 4, 5). The causes of the excess CVD risk remain unclear, but they are associated with lifestyle factors, antipsychotic use, inadequate somatic health care, and psychosocial challenges (6–8). In addition, a genetic susceptibility to CVD may play a role, as indicated by glucose disturbances in first-episode drug-naive patients with schizophrenia and increased CVD risk in first-degree relatives of individuals with schizophrenia (9, 10). However, low body mass index (BMI) has been implicated as a risk factor for schizophrenia (11). Moreover, there is considerable individual variation in other CVD risk factors, including hypertension, type 2 diabetes (T2D), and dyslipidemia (3, 8, 12), which might suggest increased genetic liability to CVD in subgroups of patients with schizophrenia.
Schizophrenia is a complex polygenic disorder with an estimated heritability of 60%–80% (13, 14). The largest genome-wide association study (GWAS) of schizophrenia to date identified 287 loci associated with the disorder (15). GWASs have also detected several loci associated with coronary artery disease (CAD) (16) and CVD risk factors, including BMI (17), waist-to-hip ratio (WHR) (18), T2D (19), total cholesterol (TC) (20), low-density lipoprotein (LDL) (20), high-density lipoprotein (HDL) (20), triglycerides (TG) (20), systolic blood pressure (SBP) (21), and diastolic blood pressure (DBP) (21). Increasing evidence suggests genetic overlap between schizophrenia and CVD risk factors (22–26). For instance, genetic variants jointly associated with increased risk of T2D and schizophrenia have been identified (23, 24). Studies have also revealed overlapping loci between schizophrenia and BMI, WHR, lipids, and SBP (22, 25, 27). Interestingly, the majority of the shared loci between schizophrenia and BMI had opposite effect directions (25), in line with a negative genetic correlation estimate (rg=−0.11) (25). However, there was an even distribution of concordant and opposite effect directions among the schizophrenia loci shared with lipids and SBP (22). Similarly, we recently discovered shared variants between bipolar disorder and CVD risk factors that had a mixture of effect directions (28). The pattern of mixed effects may indicate variation in genetic susceptibility to CVD across subgroups of patients (28, 29).
Tobacco smoking is highly prevalent among individuals with schizophrenia and is a major risk factor for CVD (3, 8). Recent data suggest a positive genetic correlation between schizophrenia and smoking (rg=0.15) (30, 31). However, genetic correlations provide an incomplete understanding of the overlapping genetic architecture between two phenotypes (14). A genetic correlation provides a summary measure between −1 and 1 of the correlation of effect sizes across all single-nucleotide polymorphisms (SNPs) (14). Thus, shared variants with a mixture of concordant and discordant effect directions cancel each other out, which may result in negligible genetic correlations (28, 32). Accordingly, genetic correlation analyses should be complemented with methods that can detect genetic overlap irrespective of effect directions (14) to identify the underlying molecular underpinnings.
Here, we aimed to further elucidate the genetic underpinnings of schizophrenia and comorbid CVD (22) by analyzing data from unprecedentedly large GWAS samples (15–21, 30), including the most recent schizophrenia GWAS (15). We applied the bivariate causal mixture model (MiXeR) (33), which estimates the total number of unique and shared genetic variants between pairs of phenotypes. We next used conditional and conjunctional false discovery rate (condFDR and conjFDR) approaches, which leverage genetic overlap between two phenotypes to boost statistical power to identify novel loci associated with a single phenotype (condFDR) and loci jointly associated with two phenotypes (conjFDR) (32). While MiXeR estimates the number of genetic variants influencing a phenotype, condFDR and conjFDR approaches discover specific genetic loci, which is crucial to obtain biological insights (22, 32). MiXeR and the conjFDR approach are able to identify genetic overlap regardless of the effect directions (32, 33).
Methods
Participant Samples
We obtained GWAS summary statistics from independent samples of schizophrenia and CVD phenotypes. GWAS results for schizophrenia were retrieved from the Psychiatric Genomics Consortium, consisting of 53,386 patients with schizophrenia and 77,258 control participants of European descent (15). We used GWAS data on CVD phenotypes, including the CVD risk factors BMI (N=795,640) (17), WHR adjusted for BMI (N=694,649) (18), T2D (N=231,426) (19), lipids (including TG, HDL, LDL, and TC) (N=1,320,016) (20), SBP and DBP (N=745,820) (21), smoking initiation (indicating whether a person has ever smoked regularly) (N=1,232,091), cigarettes per day (N=337,334) (30), and CAD (N=332,477) (16) from samples of European ancestry. All GWASs investigated in this study were approved by local ethics committees, and all participants provided written informed consent. The Regional Committee for Medical Research Ethics–South East Norway evaluated this protocol and found that no additional institutional review board approval was necessary because no individual data were used. Further details are available in the Methods section in the online supplement and the original GWAS publications (15–21, 30).
Statistical Analysis
We used the statistical tool MiXeR (33) to quantify polygenic overlap between schizophrenia and CVD phenotypes using GWAS summary statistics. MiXeR estimates the total number of shared and unique trait-influencing variants, and the results are represented with Venn diagrams (33). Additionally, MiXeR calculates the genome-wide correlations across all SNPs (rg) and the correlation of effect sizes within the shared genetic component (rgs). We also performed univariate MiXeR analyses for each phenotype, estimating the SNP-based heritability (h2SNP) and polygenicity, which is presented as the number of variants accounting for 90% of SNP heritability. Moreover, we constructed univariate quantile-quantile (Q-Q) plots for observed versus predicted GWAS p values, as well as Q-Q plots partitioned by minor allele frequency and linkage disequilibrium (LD) for each trait. These figures help inform whether the GWASs were sufficiently powered. We evaluated model fit both for univariate and bivariate analyses; that is, we evaluated the ability of the MiXeR model to predict the actual GWAS data, based on modeled versus actual conditional Q-Q plots, negative log-likelihood plots, and Akaike information criterion (AIC). In addition, we provided the Bayesian information criterion (BIC), a more stringent measure of model fit. For details, see the Methods section in the online supplement and Frei et al. (33). The MiXeR model is sensitive to LD structure estimates. In this study, LD structure was estimated using the 1000 Genomes Phase 3 genotype reference panel (34).
We constructed conditional Q-Q plots to visualize cross-trait enrichment, which is present when the proportion of SNPs associated with a phenotype (e.g., schizophrenia) increases as a function of the strength of the association with a secondary phenotype (e.g., BMI) (32). In the conditional Q-Q plots, this cross-trait enrichment is visualized as successive leftward shifts from the null distribution and can be directly interpreted in terms of the true discovery rate (1−FDR) (22, 32).
The condFDR approach was used to increase discovery of specific genetic loci associated with schizophrenia and CVD phenotypes at condFDR<0.01 (22, 32). This method uses cross-trait enrichment observed in the conditional Q-Q plots to re-rank the test statistics of a primary phenotype (e.g., schizophrenia) based on a conditional variable (e.g., BMI) (22, 32). To identify shared genetic loci, we applied a threshold of conjFDR<0.05. The conjFDR statistic is defined as the maximum of two condFDR values, providing a conservative estimate of the FDR for association with both phenotypes (32) (see the Methods section in the online supplement ).
Schizophrenia and some CVD phenotypes are strongly associated with the major histocompatibility complex (MHC), a genomic region with a complex LD pattern (14, 35). In addition, the chromosomal region 8p23.1 comprises highly correlated SNPs within a long-range LD region, and it is also associated with schizophrenia (36). Thus, we excluded the MHC and 8p23.1 regions before fitting the FDR model to avoid inflating FDR estimates, in line with previous studies (25, 28, 32). Although the conditional Q-Q plots and the FDR model are computed after excluding SNPs in these regions, we allowed for detection of SNPs within these regions because the shared genetic signal may be of high biological importance (32). For MiXeR analyses, we also excluded the MHC region as recommended (33). For additional details, see the Methods section in the online supplement .
Genomic Loci Definition
We defined independent genomic loci with FUMA, an online tool for functional mapping of genetic variants (http://fuma.ctglab.nl/) (37). Independent significant SNPs were defined as SNPs with condFDR<0.01 or conjFDR<0.05 that were independent from each other at LD r2<0.6. A subset of these that were independent from each other at r2<0.1 were considered lead SNPs. Distinct genomic loci were defined by identifying all SNPs in LD (r2≥0.6) with one of the independent significant SNPs in the locus (32). We merged all loci <250 kb apart and selected the SNP with the lowest p value as lead SNP of the merged locus. A locus was considered novel to schizophrenia if it was not physically overlapping with loci in the original GWAS (15), GWAS catalog, or other relevant studies (see the Methods section in the online supplement ).
Effect Directions and Genetic Correlations
We evaluated the directional effects of the shared loci between schizophrenia and CVD phenotypes by comparing their z scores. Genetic correlations were estimated using MiXeR and LD score regression and were corrected for multiple testing (38).
Validation of conjFDR Results
To test the validity of the findings, we assessed the consistency in allelic effect directions of shared lead SNPs between the discovery data sets and independent data sets (i.e., sign concordance), in line with previous studies (39, 40). Since the probability of replicating individual loci at genome-wide significance is low in smaller samples because of weak genetic effects, we identified lead SNPs (or the next most significant candidate SNPs if the lead SNP was not present in the replication sample) that were nominally significant at p<0.05 in each replication sample. This approach has been applied previously, including in several high-profile GWASs, such as GWASs of educational attainment and intelligence (41, 42), and in condFDR and conjFDR studies (39, 40, 43, 44). The replication data for schizophrenia (15) and CVD phenotypes (20, 45–48) were retrieved from East Asian samples (see the Methods section in the online supplement ).
Functional Annotation
Using FUMA (37), we functionally annotated candidate SNPs within the genomic loci, defined as any SNP with a condFDR or conjFDR value<0.10 and an r2≥0.6 with one of the independent significant SNPs. SNPs were annotated with the combined annotation-dependent depletion score (49), a method that predicts the deleteriousness of SNPs on protein structure or function; RegulomeDB scores (50), which predict regulatory functions; and chromatin states that indicate the transcription or regulation effects at the SNP locus (51, 52). Next, candidate SNPs were mapped to putative causal genes, using three strategies: positional mapping (physical proximity), expression quantitative trait locus (eQTL) mapping, and chromatin interaction mapping (37). This three-strategy approach has high sensitivity but is liable to false positives that influence gene-set enrichment analyses. Thus, to reduce false positives, we performed gene-set and pathway analyses of genes nearest to lead SNPs (37). Studies suggest that the nearest gene is often the causal gene (53), although this is not always the case (54).
We also conducted supplementary gene-set analyses of the genes mapped to candidate SNPs using the same three strategies (37). We excluded mapped genes in MHC and 8p23.1 before these enrichment analyses to limit the inflation that may result from the high LD patterns characterizing these regions. As several correlated SNPs reside in these regions, single associations in these clusters can drive the apparent enrichment of entire gene sets. However, we also performed gene-set and pathway analyses of mapped genes included in MHC and 8p23.1 to check whether there were considerable differences in findings from analyses with versus those without these genomic regions. For details, see the Methods section in the online supplement .
Genetic Overlap Between CVD Phenotypes and Genomic Structural Equation Modeling
We also used LD score regression and MiXeR analyses to assess genetic overlap between the CVD phenotypes to aid in the interpretation of the schizophrenia and CVD results (see the Methods section in the online supplement ). In addition, we investigated the influence of CVD risk factors on the relationship between schizophrenia and CAD by performing genomic structural equation modeling analyses that included multiple regression (partial correlation) and mediation analyses (55, 56).
The data from the condFDR, conjFDR, replication, and functional analyses are presented in Tables S1–S98, the genetic correlations between CVD traits are presented in Table S99, and the MiXeR data are presented in Tables S100–S102 in the online supplement .
Results
Genetic Overlap Between Schizophrenia and CVD Phenotypes
The univariate MiXeR results are presented in Table S100 in the online supplement . The analyses revealed large differences in the number of variants accounting for 90% of SNP heritability across phenotypes: smoking initiation (N=11.1K), BMI (N=11.0K), and schizophrenia (N=9.6K) demonstrated the highest polygenicity, whereas SBP and DBP (N=4.4K and 3.9K), T2D (N=2.3K), lipids (N=0.8K to 1.8K), WHR (N=1.7K), and CAD (N=1.3K) showed lower polygenicity (Figure 1; see also Table S100 in the online supplement ). SNP-based heritability, univariate Q-Q plots for observed versus predicted GWAS p values, and Q-Q plots partitioned by minor allele frequency and LD for each trait are presented in Figures S1–S2 and described in the Results section in the online supplement . The univariate AIC and BIC values were all highly positive (see Table S100 in the online supplement ), indicating sufficient model fit.
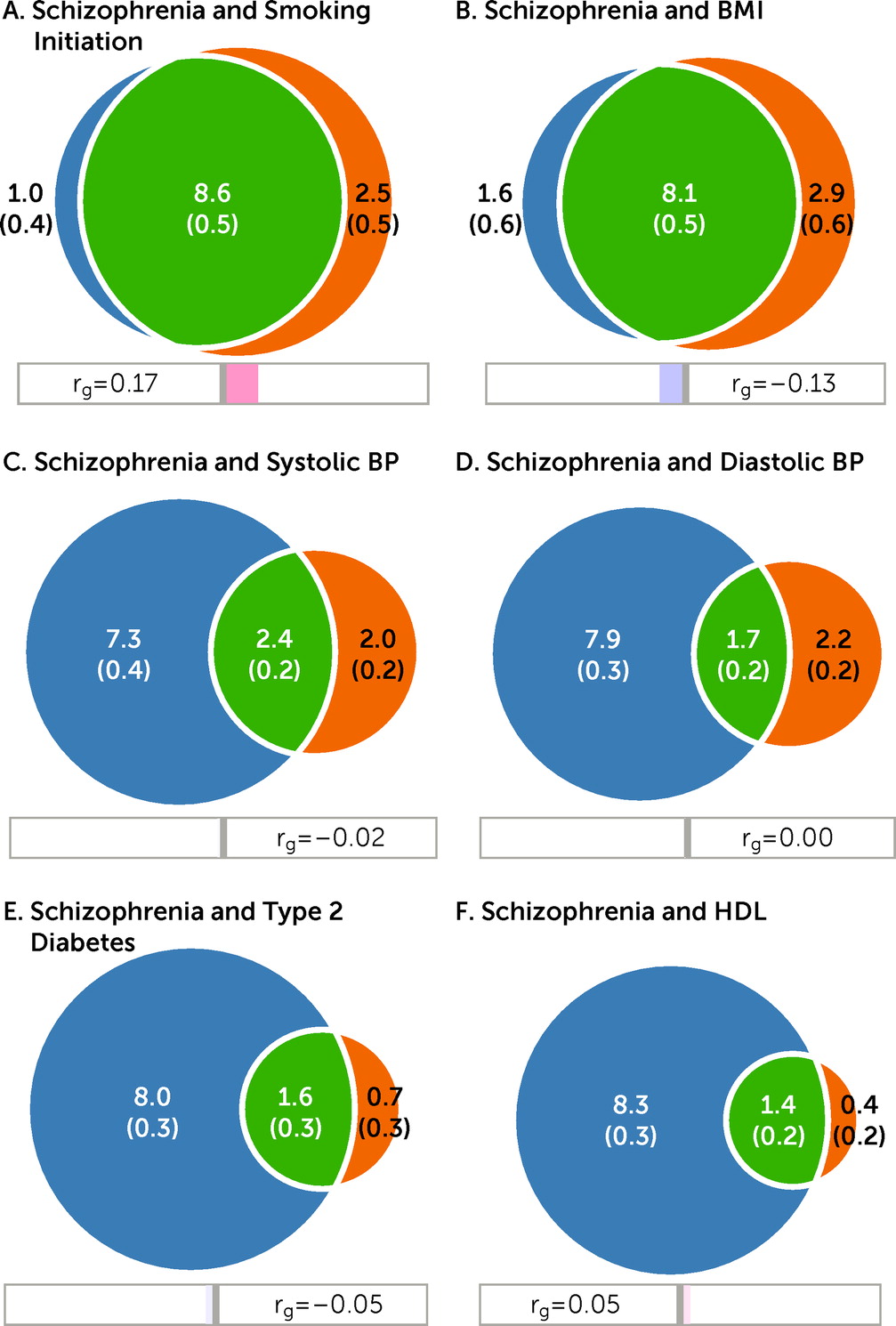
FIGURE 1. Shared and unique polygenic components between schizophrenia and cardiovascular disease phenotypesa
aThe Venn diagrams illustrate shared and unique trait-influencing variants, showing polygenic overlap (green) between schizophrenia (blue) and cardiovascular disease phenotypes (orange), including A) smoking initiation, B) body mass index (BMI), C) systolic blood pressure (BP), D) diastolic BP, E) type 2 diabetes, and F) high-density lipoprotein (HDL). The numbers in the Venn diagram indicate the estimated quantity of shared and unique trait-influencing variants (in thousands), explaining 90% of single-nucleotide polymorphism heritability in each phenotype, and the standard error. The size of the circles reflects the degree of polygenicity. The genetic correlations (rg) are also provided. The figures are based on MiXeR analysis.
The bivariate analyses indicated substantial genetic overlap between schizophrenia and some CVD phenotypes (Figure 1). In particular, MiXeR estimated that of the 9.6K schizophrenia-influencing variants, 8.6K and 8.1K also influence smoking initiation and BMI, respectively (Figure 1A–B). This corresponds to approximately 90% and 84% of schizophrenia-influencing variants overlapping with smoking initiation and BMI, respectively (Table 1). Schizophrenia shared fewer variants with SBP (N=2.4K); DBP (N=1.7K); T2D (N=1.6K); lipids such as HDL (N=1.4K) (Figure 1C–F), TG (N=1.2K), LDL (N=0.3K), and TC (N=0.3K); WHR (N=1.2K); and CAD (N=0.5K). These findings were in part because of lower polygenicity of these CVD phenotypes (see Figures S3–S10 in the online supplement ). Bivariate MiXeR was deemed to model the data adequately, as indicated by Q-Q plots, log-likelihood curves (see Figures S3–S10 in the online supplement ), and AIC values (see Table S101 in the online supplement ). The BIC values, which provide a stricter index of model fit than AIC values, were lower, but generally positive, indicating adequate model fit (see Table S101 in the online supplement ). MiXeR was not able to model cigarettes per day adequately, as shown in the log-likelihood curve in Figure S11 in the online supplement . See the Results section in the online supplement for further details.
| Cardiovascular Disease Trait | Schizophrenia-Related Variants Shared With Cardiovascular Disease | Cardiovascular Disease–Related Variants Shared With Schizophrenia | Genetic Correlation of Shared Variants | |
|---|---|---|---|---|
| | % | % | Mean | SD |
| Smoking initiation | 89.6 | 77.5 | 0.20 | 0.01 |
| BMI | 83.5 | 73.6 | −0.17 | 0.01 |
| Systolic BP | 24.7 | 54.5 | −0.04 | 0.01 |
| Diastolic BP | 17.7 | 43.6 | 0.01 | 0.01 |
| Type 2 diabetes | 16.7 | 69.6 | −0.13 | 0.03 |
| HDL | 14.4 | 77.8 | 0.15 | 0.01 |
| Triglycerides | 12.4 | 85.7 | −0.06 | 0.02 |
| LDL | 3.1 | 37.5 | 0.30 | 0.11 |
| Total cholesterol | 3.1 | 33.3 | 0.40 | 0.17 |
| Coronary heart disease | 5.2 | 34.5 | 0.17 | 0.03 |
| Waist-to-hip ratio | 12.12 | 43.53 | 0.29 | 0.08 |
a
The table lists the proportions of schizophrenia-influencing variants shared with each cardiovascular disease phenotype and vice versa. Genetic correlation of shared variants: genetic correlation of effect sizes within the shared genetic component. The variants explain 90% of single-nucleotide polymorphism heritability of each phenotype. The waist-to-hip ratios were adjusted for BMI. BMI=body mass index; BP=blood pressure; HDL=high-density lipoprotein; LDL=low-density lipoprotein.
The genetic correlations within the shared component (green areas in the Venn diagrams, Figure 1) are presented in Table 1. MiXeR estimated moderate positive genetic correlations within the shared component for schizophrenia and smoking initiation (rgs=0.20), cigarettes per day (rgs=0.36), LDL (rgs=0.30), TC (rgs=0.40), HDL (rgs=0.15), CAD (rgs=0.17), and WHR (rgs=0.29), whereas negative genetic correlations were estimated within the shared component between schizophrenia and BMI (rgs=−0.17) and T2D (rgs=−0.13); the remaining correlations were close to zero (Table 1). The genome-wide correlations (Table 2) largely mirrored the correlations within the shared components (Table 1), yet they were lower and nonsignificant for lipids, CAD, T2D, and WHR after correction for multiple testing.
| Associated Phenotypes | Shared Loci | Concordant Effect | Genetic Correlation | |
|---|---|---|---|---|
| | N | % | rg | p |
| BMI | 304 | 34.5 | −0.112 | 8.04×10−13 |
| Waist-to-hip ratio | 193 | 52.3 | 0.039 | 0.028 |
| Smoking initiation | 293 | 68.9 | 0.158 | 4.36×10−13 |
| Cigarettes per day | 129 | 62.8 | 0.137 | 2.67×10−7 |
| Systolic BP | 294 | 47.6 | −0.004 | 0.817 |
| Diastolic BP | 259 | 47.9 | 0.004 | 0.823 |
| Type 2 diabetes | 147 | 46.9 | −0.053 | 0.007 |
| Triglycerides | 307 | 47.2 | −0.037 | 0.002 |
| HDL | 304 | 55.6 | 0.058 | 0.003 |
| LDL | 158 | 51.3 | 0.029 | 0.090 |
| Total cholesterol | 176 | 53.5 | 0.034 | 0.042 |
| Coronary heart disease | 35 | 54.3 | −0.021 | 0.471 |
a
The table lists the number of shared loci at conjunctional false discovery rate <0.05, percentage of loci with concordant effect directions, and genetic correlation (rg) estimated by linkage disequilibrium score regression. BMI=body mass index; BP=blood pressure; HDL=high-density lipoprotein; LDL=low-density lipoprotein.
Loci Shared Between Schizophrenia and CVD Phenotypes
After observing cross-trait enrichment in conditional Q-Q plots (see Figures S12–S13 in the online supplement ), we applied a condFDR approach that identified several loci associated with schizophrenia conditional on CVD phenotypes (see Tables S1–S12 in the online supplement ), including smoking initiation (N=362), SBP (N=325), DBP (N=317), TG (N=332), HDL (N=331), TC (N=272), LDL (N=279), cigarettes per day (N=307), WHR (N=303), and BMI (N=299). Next, we discovered multiple loci associated with CVD phenotypes conditional on schizophrenia (see Tables S13–S24 in the online supplement ).
A total of 825 distinct loci were jointly associated with schizophrenia and CVD phenotypes at conjFDR<0.05 (Figure 2 and Table 2; see also Tables S25–S36 in the online supplement ). Schizophrenia shared 304 loci with BMI, 193 with WHR, 293 loci with smoking initiation, 129 loci with cigarettes per day, 307 with TG, 304 with HDL, 176 with TC, 158 with LDL, 294 with SBP, 259 with DBP, 147 with T2D, and 35 with CAD (Figure 2 and Table 2; see also Tables S25–S36 in the online supplement ). In addition, 357 loci were common for schizophrenia and more than one CVD phenotype (see Table S37 and Figure S14 in the online supplement ).
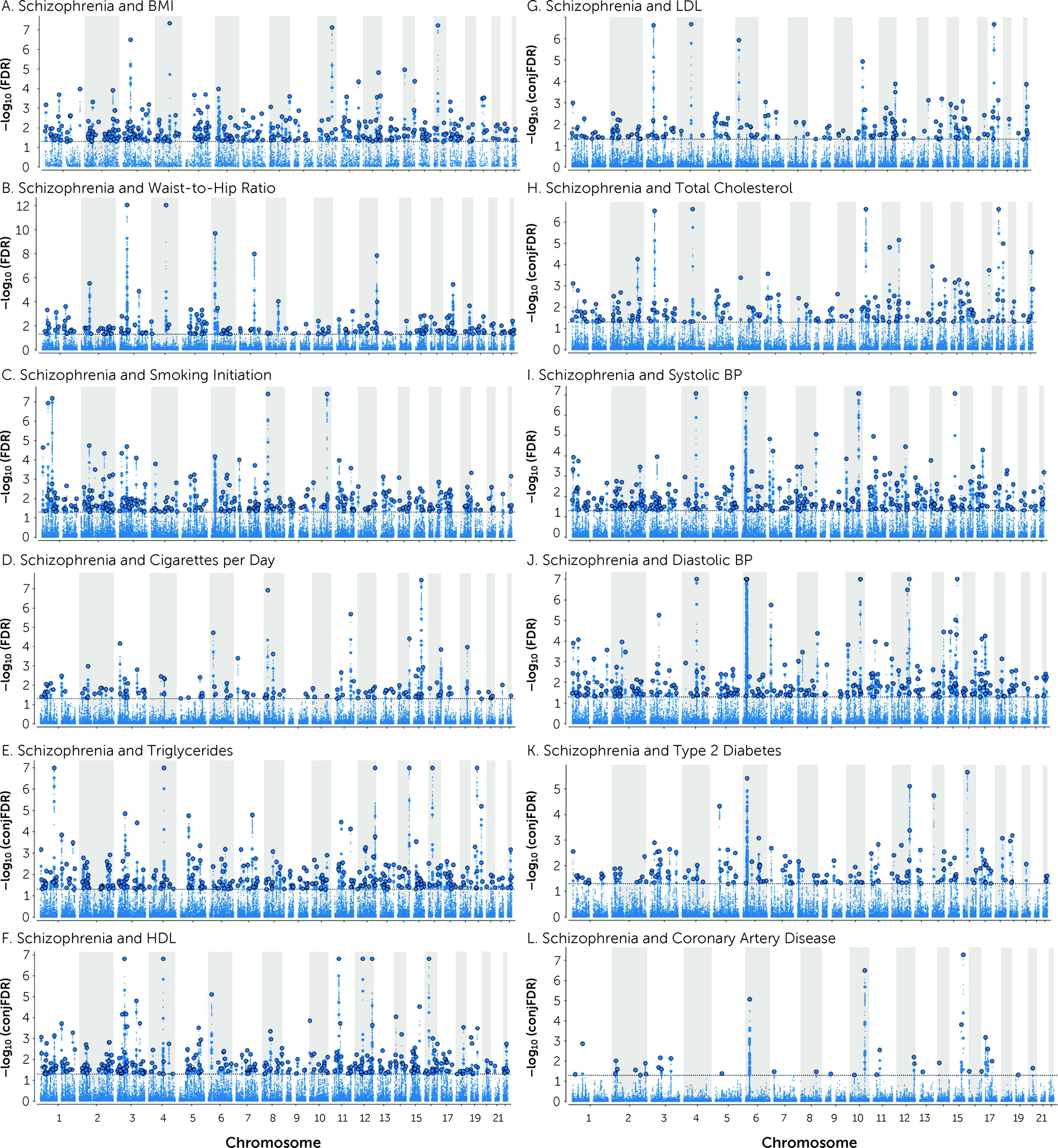
FIGURE 2. Common genetic variants jointly associated with schizophrenia and cardiovascular disease phenotypesa
aThe Manhattan plots show the common genetic variants jointly associated with schizophrenia and A) body mass index (BMI), B) waist-to-hip ratio (adjusted for BMI), C) smoking initiation, D) cigarettes per day, E) triglycerides, F) high-density lipoprotein (HDL), G) low-density lipoprotein (LDL), H) total cholesterol, I) systolic blood pressure (BP), J) diastolic BP, K) type 2 diabetes, and L) coronary artery disease at conjunctional false discovery rate (conjFDR)<0.05. Each panel shows the –log10 transformed conjFDR values for each single-nucleotide polymorphism (SNP) on the y-axis and chromosomal positions along the x-axis. SNPs with conjFDR<0.05 (i.e., −log10 FDR>1.3) are shown with enlarged data points. A black circle around the enlarged data points indicates the most significant SNP in each linkage disequilibrium block. The figure shows the localization of the “conjunctional loci”; additional details are provided in the tables in the online supplement .
Of the loci associated with schizophrenia at condFDR<0.01, 104 are novel schizophrenia loci (see Table S38 in the online supplement ). Of the shared loci at conjFDR<0.05, 348 are novel for schizophrenia (see Table S39 in the online supplement ). This yielded a total of 366 novel schizophrenia loci with a condFDR<0.01 or conjFDR<0.05 (see Table S40 in the online supplement ).
We determined the directionality of effects of lead SNPs within shared loci (Table 2; see also Tables S25–S36 in the online supplement ): 35% were concordant for schizophrenia and BMI, 69% were concordant for schizophrenia and smoking initiation, and 63% were concordant for schizophrenia and cigarettes per day. Approximately half of the loci shared between schizophrenia and lipids, SBP, DBP, T2D, WHR, and CAD possessed concordant effect directions (Table 2; see also Tables S25–S36 in the online supplement ). The effect directions broadly reflected the directions observed using genetic correlations (Table 1 and Table 2).
Validation: Consistency of Genetic Effects in Independent Samples
Validation analyses demonstrated a high degree of sign concordance (∼78%) between the independent discovery and replication data sets for the shared loci at conjFDR<0.05 (see Tables S41–S47 in the online supplement ). For exact binomial values, see the Results section in the online supplement . The replication GWASs were considerably smaller than the discovery samples, which reduced the power to detect significant lead SNPs. Nevertheless, the consistency of associations in the replication data sets was comparable to that of other GWASs of complex traits (39, 40), supporting the validity of these findings. Of the 304 lead SNPs in loci shared between schizophrenia and BMI, 73 and 74 had p values <0.05 in the replication samples for schizophrenia and BMI, respectively. Of the 294 lead SNPs in loci shared between schizophrenia and SBP, 58 and 91 had p values <0.05 in the replication samples, respectively. Of the 293 lead SNPs in loci shared between schizophrenia and smoking initiation, 44 and 40 had p values <0.05 in the replication samples, respectively. Of the 129 lead SNPs in loci shared between schizophrenia and cigarettes per day, 26 and 16 had p values <0.05 in the replication samples, respectively. Of the 307 lead SNPs in loci shared between schizophrenia and TG, 57 and 41 had p values <0.05 in the replication samples, respectively. Of the 304 lead SNPs in loci shared between schizophrenia and HDL, 50 and 41 had p values <0.05 in the replication samples, respectively. Of the 35 lead SNPs in loci shared between schizophrenia and CAD, nine and three had p values <0.05 in the replication samples, respectively. See the Results section and Tables S41–S47 in the online supplement for more information.
Functional Annotation
Functional annotation of candidate SNPs shared between schizophrenia and CVD phenotypes demonstrated that they were mostly intronic and intergenic (see Tables S48–S59 in the online supplement ). Next, we mapped candidate SNPs to genes using three gene-mapping strategies and discovered thousands of protein-coding genes (see Tables S60–S71 in the online supplement ). Several SNPs were mapped to genes with chromatin interaction and eQTL associations in human brain tissue (e.g., fetal and adult cortex) and cells of the immune system (e.g., monocytes and CD4 T cells) (see the Results section and Tables S60–S71 in the online supplement ).
The gene-set analysis of genes nearest to lead SNPs shared between schizophrenia and BMI implicated enrichment in several Gene Ontology (GO) terms, the most strongly associated terms being “regulation of transmembrane transport,” “regulation of synaptic plasticity,” and several neuronal gene sets (see Table S72 in the online supplement ). There were no significant GO terms enriched among the genes nearest to lead SNPs shared with WHR, except a significant pathway termed “KEGG Alzheimer’s disease” (see Table S73 in the online supplement ). Gene-set analysis of the genes nearest to the lead SNPs shared between schizophrenia and smoking initiation indicated GO terms with a predominance of gene sets related to neurodevelopment, including “central nervous system development” (see Table S74 in the online supplement ). There were also overrepresented pathways among these genes, including “pathways affected in adenoid cystic carcinoma” (see Table S74 in the online supplement ). The enriched gene sets associated with schizophrenia and lipids, SBP, and DBP involved neuronal, synaptic, and immunological gene sets (see Tables S75–S79 in the online supplement ). In addition, there were significantly enriched gene sets associated with DNA-binding and cellular processes (see Tables S75–S79 in the online supplement ). Pathway analysis of the genes mapped to schizophrenia and TG, SBP, and DBP indicated “MAPK signaling pathway,” “brain-derived neurotrophic factor signaling pathway,” and “energy metabolism” (see Tables S75, S78, and S79 in the online supplement ). Moreover, the enrichment analyses including genes in MHC and 8p23.1 yielded results similar to those excluding these genomic regions (see Tables S72–S79 in the online supplement ). This indicates that these genomic regions do not have a substantial impact on the results.
We also performed gene-set and pathway analyses with genes mapped to all candidate SNPs. The results agreed broadly with the results based on genes nearest to lead SNPs, yet were less specific and included several intra- and intercellular gene sets (see the Results section and Tables S80–S98 in the online supplement ).
Genetic Overlap Between CVD Phenotypes
The genetic correlations between the CVD phenotypes ranged from low to high, with the strongest correlations between TC and LDL (rg=0.946, p=3.35×10−12) and between SBP and DBP (rg=0.808, p<1.00×10−20) and the weakest between lipids and blood pressure (rg=−0.01 to −0.12, p=0.62 to 1.54×10−9) (see Figure S15 and Table S99 in the online supplement ). MiXeR analyses demonstrated different levels of genetic overlap, with the most overlap between SBP and TG, BMI and CAD, BMI and TG, and BMI and T2D (see Figure S16 in the online supplement ). Adequate MiXeR model fit, however, was restricted to BMI and smoking initiation, CAD and TG, SBP and TG, CAD and SBP, SBP and smoking initiation, SBP and T2D, and BMI and SBP (see Figure S16 and Table S102 in the online supplement ). The other bivariate estimates were uncertain, as indicated by no clear minimum on the log-likelihood curves, Q-Q plots (the observed Q-Q plots did not closely follow the model predictions), and negative AIC and BIC values (see Figure S16 and Table S102 in the online supplement ).
Genomic Structural Equation Modeling Results
We further explored the genetic relationship between schizophrenia and CAD controlling for the effect of risk factors (smoking initiation, BMI, HDL, SBP, and T2D). The multiple regression analyses demonstrated no statistically significant impact of these CVD risk factors, except for a minor effect of smoking initiation on the relationship between schizophrenia and CAD, resulting in a slightly modified genetic correlation between schizophrenia and CAD (rg=−0.055, p=0.041) (see Figure S17 in the online supplement ). Mediation analyses, however, found statistically significant effects for all CVD risk factors except SBP (see Figure S18 in the online supplement ), although the indirect effect between schizophrenia and CAD remained close to zero (rg=−0.035 to 0.036), indicating minor effects. For further details, see the Results section and Figures S17–S18 in the online supplement .
Discussion
In this study, we found that a considerable proportion of genetic variants underlying schizophrenia also influence CVD phenotypes, particularly the risk factors smoking initiation and BMI, using MiXeR. Next, we detected more than 800 distinct loci jointly associated with schizophrenia and CVD risk factors and CAD at conjFDR<0.05. Most of the loci shared between schizophrenia and smoking initiation and cigarettes per day possessed concordant effect directions, in line with positive genetic correlations. The overlapping loci with BMI had mainly opposite effect directions, consistent with negative genetic correlations. There was a pattern of mixed effect directions among loci jointly associated with schizophrenia and the other CVD phenotypes, including lipids, SBP, DBP, T2D, WHR, and CAD. The high degree of sign concordance between the discovery and replication samples supports the validity of the findings. Furthermore, functional analyses of the shared loci implicated genes associated with neurodevelopment and the immune system. These results shed light on putative biological functions and pathways associated with the comorbidity between CVD and schizophrenia that warrant further investigation and experimental validation.
The MiXeR analyses indicated differences in the level of genetic overlap across various CVD risk factors and CAD. In particular, MiXeR estimated that the majority of the SNPs influencing schizophrenia also affect smoking initiation (∼90%) and BMI (∼84%), whereas less genetic overlap was observed with the other CVD risk factors. Smoking and BMI seem to be more polygenic than the other CVD phenotypes. The causes of the differences in polygenicity are unclear but may be related to smoking and BMI being more influenced by behavior that is regulated by the brain than the other CVD phenotypes. Other analyses have also revealed that behavioral phenotypes have higher polygenicity than somatic diseases and traits (57). Lower polygenicity of the other CVD phenotypes compared with BMI and smoking initiation limits the genetic overlap with schizophrenia.
Our conjFDR analyses detected several genetic loci jointly associated with schizophrenia and CVD risk factors, especially BMI, smoking initiation, blood pressure, and TG. Larger GWAS samples of these CVD phenotypes compared with the GWASs of T2D and CAD are likely to contribute to the greater number of identified loci. A total of 366 of the identified loci are novel to schizophrenia. The results illustrate the increased SNP discovery by leveraging pleiotropy with CVD by using the condFDR and conjFDR approaches (22, 32). The allelic effect directions of the shared loci identified by conjFDR were mainly in line with the genome-wide correlation and genetic correlations within the shared components obtained from MiXeR. Note, however, that the identified loci at conjFDR<0.05 represent only a small fraction of the genetic architecture, and the concordance in allelic effects among these shared loci may not necessarily align with the measures of genetic correlation at the genome-wide level or within the shared components.
Furthermore, the majority of the shared loci have opposite allelic effect directions in schizophrenia and BMI. The results indicate that people with schizophrenia are genetically predisposed to lower BMI, at the group level. This is in line with previous genetic findings (25) and clinical evidence of low BMI being a risk factor for schizophrenia (11) and being more prevalent among individuals with schizophrenia than in the general population (58, 59). However, obesity is also more common in individuals with schizophrenia compared with those in the general population (3, 58). The present findings indicate that factors other than common genetic variants play an important role in weight gain in schizophrenia (25). In particular, adverse effects of antipsychotics and unhealthy lifestyle related to negative symptoms, depression, and socioeconomic challenges are likely to be major causes (6–8, 60, 61). However, genetic factors appear to play an important role in antipsychotic-induced weight gain as well, with a heritability estimate of 60%–80% (62). Taken together, the evidence indicates interactions between genetic liability to antipsychotic-induced weight gain, lifestyle factors, and psychosocial challenges as underlying mechanisms of weight gain.
Despite the negative genetic correlation between BMI and schizophrenia, a trend toward a positive association was observed for WHR (rg=0.039). Although this may seem inconsistent with the inverse schizophrenia-BMI association, WHR correlates weakly with BMI clinically (63), and WHR adjusted for BMI demonstrated no significant genetic correlation with BMI in our study (see Table S99 in the online supplement ). Moreover, the genetic correlation between schizophrenia and WHR was not significant after correction for multiple testing, in line with previous studies (27, 64), although one study found a statistically significant, yet low, negative genetic correlation between schizophrenia and waist circumference (rg=−0.07) (65). Furthermore, the results indicate that the higher WHR seen in antipsychotic-naive patients with schizophrenia (66) is not due mainly to genetic factors, but rather to lifestyle factors.
We discovered extensive genetic overlap between schizophrenia and smoking behavior. The effect directions of the shared SNPs are in line with the moderate positive genetic correlation estimated here and in previous studies (30, 31). The results indicate an increased genetic propensity for smoking associated with schizophrenia. The addictive properties of nicotine may have a larger influence in people with schizophrenia, in line with evidence of greater nicotine dependence among individuals with schizophrenia than in the general population (67). In particular, patients with schizophrenia experience greater reinforcing effects of nicotine and more severe withdrawal symptoms during abstinence (67, 68). The higher nicotine dependence in schizophrenia may be partly genetically driven, as indicated by a positive genetic correlation (69). Moreover, studies have linked both nicotine dependence and schizophrenia to variants in the nicotinic acetylcholine receptor (nAChR) gene cluster (CHRNA3-CHRNA5-CHRNB4) (15, 69). Here, we corroborated associations of variants in the nAChR gene cluster with schizophrenia and cigarettes per day (see Table S51 in the online supplement ). In addition, smoking may represent a form of self-medication. Nicotine activates nAChRs, which stimulates release of dopamine, serotonin, and glutamate (68). Thus, tobacco smoking in people with schizophrenia may involve, to some extent, an attempt to compensate for genetically determined dysfunction of nAChRs, implicating monoaminergic and glutamatergic signaling (14, 68), although this requires further investigation.
We identified several shared loci with mixed effect directions in schizophrenia and lipids, SBP, DBP, WHR, T2D, and CAD. The results validate earlier findings of bidirectional effects among overlapping loci between schizophrenia and CVD risk factors (22, 26, 27) and between bipolar disorder and CVD risk factors (28). Moreover, the present study extends previous results (22) by providing a more detailed characterization of the shared genetic architecture and specific overlapping loci. Nevertheless, the common SNPs identified here and previously (22, 26–28) have small effects, explaining a small portion of the overall disease risk. The remaining variance is likely to be explained by multiple undetected SNPs, rare variants, interactions between genes, and interactions between genes and environmental factors, especially antipsychotics and unhealthy lifestyle (6, 60).
Although the average level of CVD risk is higher in schizophrenia compared with the general population, there is considerable individual variation in CVD risk (3, 8, 12). Our findings of shared loci with mixed effect directions may reflect variation in genetic susceptibility to CVD across subgroups of schizophrenia. Thus, there may be subsets of patients with a higher genetic liability to CVD, as indicated by previous studies (29, 70). This possibility can help explain reports of cardiometabolic disturbances in drug-naive patients with schizophrenia at illness onset and among their relatives compared with healthy control participants (9, 10, 71). Moreover, a recent study provided evidence of genetic contribution to T2D in schizophrenia (24), although findings have been inconsistent (15, 72). Patients with more negative symptoms may represent a subgroup who are at greater risk for CVD, consistent with findings suggesting that patients with more severe and enduring negative symptoms have a higher CVD risk, whereas positive psychotic symptoms seem to be less associated with CVD (73, 74). Larger GWAS samples with phenotypic refinement are required to identify potential subgroups of schizophrenia patients with differential liability to CVD.
Both overweight and smoking are risk factors for CAD, in line with their positive genetic correlation with CAD shown here. We investigated whether BMI and smoking initiation, which have a negative and positive genetic correlation with schizophrenia, respectively, influence the genetic relationship between schizophrenia and CAD. We hypothesized that controlling for these CVD risk factors would modify the association between schizophrenia and CAD. The results from the genomic structural equation modeling suggested minimal effect on the relationship between schizophrenia and CAD, indicating that there are other factors explaining the nonsignificant correlation between them. Furthermore, we did not find support for a systematic change in the direction of the association between schizophrenia and CAD by controlling for the other CVD risk factors. These findings support a mixture of effect directions across the different CVD risk factors. Thus, it does not appear as though controlling for the effect of a CVD factor (e.g., smoking) affects a subgroup of variants that are shared with schizophrenia and CAD. However, the statistical power for finding such a pattern may have been inadequate.
Functional analyses of the shared loci implicated genes associated with neurodevelopment, synaptic function, immune system, intra- and intercellular processes, and metabolic mechanisms. The results are in line with the neurodevelopmental hypothesis and with the immune system being implicated in the pathogenesis of schizophrenia (14). In addition, brain function regulates behavior, which plays a key role in lifestyle habits influencing CVD. The immune system also plays a crucial role in the development of CVD (75). The findings from the functional analyses align with recent studies of genetic overlap between schizophrenia, bipolar disorder, and CVD risk factors (25, 28, 64). However, the results should be considered with caution given the limitations of functional annotation methods (37) and the complex mechanisms underlying schizophrenia and comorbid CVD. The methods used in the present study are limited by uncertainties in translating genetic loci to causal variants, which restricts the biological interpretation of the shared genetic variants (14). Some additional methodological limitations should be noted. The model underlying MiXeR is sensitive to LD structure estimates. Discrepancies between the LD structure of the samples used for the GWAS and that of the reference panel may have biased the model’s estimates. Additionally, implementation of the MiXeR model assumes similar LD structure in the analyzed GWAS, which hinders the analysis of GWASs based on genetically dissimilar populations, including transancestry analyses. Thus, we are working on implementation that supports transancestry analyses, permitting transferability of results across ethnicities.
In summary, we revealed polygenic overlap between schizophrenia and CVD risk factors, particularly BMI and smoking. The results indicate an inherent propensity to smoking in individuals with schizophrenia. In contrast, several schizophrenia risk loci appear to be protective against obesity. The findings highlight the importance of environmental factors in the development of obesity and other CVD comorbidities. In addition, the mixed effect directions of the shared loci between schizophrenia and lipids, blood pressure, T2D, and CAD may suggest variation in genetic vulnerability to CVD across schizophrenia subgroups, which may underlie some of the observed heterogeneity in CVD comorbidity. Studies with larger GWASs will uncover more of the genetic architecture of schizophrenia and may reveal differences in genetic liability to CVD across subsets of patients. Such findings could provide clinically useful discoveries that pave the way for risk stratification and more tailored interventions. Further work is needed to identify the causal genetic variants and determine their functional properties. This research is likely to provide insights into the mechanisms underlying the comorbidity and might facilitate the development of antipsychotics with lower metabolic side effects. Such progress will enable more effective prevention of comorbid CVD, thereby helping to mitigate a major clinical and health care problem.
Acknowledgments
The authors thank the research participants contributing to the genome-wide association studies (GWASs) used for the present study, including the GWASs of schizophrenia from Psychiatric Genomics Consortium and the GWASs of coronary artery disease and cardiovascular disease risk factors. This research was conducted using data from the UK Biobank, a major biomedical database, under accession number 27412 (www.ukbiobank.ac.uk). This work was performed on the Tjeneste for Sensitive Data (TSD) platform, owned by the University of Oslo and operated and developed by the TSD service group at the University of Oslo, IT-Department (tsd-drift@usit.uio.no).
Footnotes
Data availability: The data sets analyzed for this study are available in repositories of GWASs for schizophrenia (https://www.med.unc.edu/pgc/download-results/); body mass index (Genetic Investigation of Anthropometric Traits [GIANT] consortium data files) (https://portals.broadinstitute.org/collaboration/giant/index.php/); lipids (triglycerides, total cholesterol, low-density lipoprotein, and high-density lipoprotein) (http://csg.sph.umich.edu/willer/public/glgc-lipids2021/); type 2 diabetes (https://diagram-consortium.org/downloads.html); systolic and diastolic blood pressure (https://www.ebi.ac.uk/gwas/publications/30224653); coronary artery disease (http://www.cardiogramplusc4d.org/data-downloads/); smoking characteristics (https://conservancy.umn.edu/handle/11299/201564); and waist-to-hip ratio (https://github.com/lindgrengroup/fatdistnGWAS).
Code availability: The codes used for carrying out the described analyses are available at https://github.com/precimed/pleiofdr, https://github.com/precimed/mixer, and https://github.com/bulik/ldsc.
Supplementary Material
File (appi.ajp.20220660.ds001.xls)
- Download
- 28.47 MB
File (appi.ajp.20220660.ds002.pdf)
- View/Download
- 5.87 MB
References
1.
Heiberg IH, Jacobsen BK, Nesvåg R, et al: Total and cause-specific standardized mortality ratios in patients with schizophrenia and/or substance use disorder. PLoS One 2018; 13:e0202028
2.
Laursen TM, Plana-Ripoll O, Andersen PK, et al: Cause-specific life years lost among persons diagnosed with schizophrenia: is it getting better or worse? Schizophr Res 2019; 206:284–290
3.
Rødevand L, Steen NE, Elvsåshagen T, et al: Cardiovascular risk remains high in schizophrenia with modest improvements in bipolar disorder during past decade. Acta Psychiatr Scand 2019; 139:348–360
4.
Plana-Ripoll O, Weye N, Momen NC, et al: Changes over time in the differential mortality gap in individuals with mental disorders. JAMA Psychiatry 2020; 77:648–650
5.
Lambert AM, Parretti HM, Pearce E, et al: Temporal trends in associations between severe mental illness and risk of cardiovascular disease: a systematic review and meta-analysis. PLoS Med 2022; 19:e1003960
6.
Correll CU, Detraux J, De Lepeleire J, et al: Effects of antipsychotics, antidepressants and mood stabilizers on risk for physical diseases in people with schizophrenia, depression and bipolar disorder. World Psychiatry 2015; 14:119–136
7.
Badcock JC, Mackinnon A, Waterreus A, et al: Loneliness in psychotic illness and its association with cardiometabolic disorders. Schizophr Res 2019; 204:90–95
8.
De Hert M, Correll CU, Bobes J, et al: Physical illness in patients with severe mental disorders. I. prevalence, impact of medications and disparities in health care. World Psychiatry 2011; 10:52–77
9.
Mothi SS, Tandon N, Padmanabhan J, et al: Increased cardiometabolic dysfunction in first-degree relatives of patients with psychotic disorders. Schizophr Res 2015; 165:103–107
10.
Yang W, Zheng L, Zheng B, et al: A meta-analysis of abnormal glucose metabolism in first-episode drug-naive schizophrenia. Psychiatr Danub 2020; 32:46–54
11.
Zammit S, Rasmussen F, Farahmand B, et al: Height and body mass index in young adulthood and risk of schizophrenia: a longitudinal study of 1 347 520 Swedish men. Acta Psychiatr Scand 2007; 116:378–385
12.
Pérez-Piñar M, Mathur R, Foguet Q, et al: Cardiovascular risk factors among patients with schizophrenia, bipolar, depressive, anxiety, and personality disorders. Eur Psychiatry 2016; 35:8–15
13.
Sullivan PF, Kendler KS, Neale MC: Schizophrenia as a complex trait: evidence from a meta-analysis of twin studies. Arch Gen Psychiatry 2003; 60:1187–1192
14.
Smeland OB, Frei O, Dale AM, et al: The polygenic architecture of schizophrenia: rethinking pathogenesis and nosology. Nat Rev Neurol 2020; 16:366–379
15.
Trubetskoy V, Pardiñas AF, Qi T, et al: Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature 2022; 604:502–508
16.
Nelson CP, Goel A, Butterworth AS, et al: Association analyses based on false discovery rate implicate new loci for coronary artery disease. Nat Genet 2017; 49:1385–1391
17.
Turcot V, Lu Y, Highland HM, et al: Protein-altering variants associated with body mass index implicate pathways that control energy intake and expenditure in obesity. Nat Genet 2018; 50:26–41
18.
Pulit SL, Stoneman C, Morris AP, et al: Meta-analysis of genome-wide association studies for body fat distribution in 694 649 individuals of European ancestry. Hum Mol Genet 2019; 28:166–174
19.
Mahajan A, Taliun D, Thurner M, et al: Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat Genet 2018; 50:1505–1513
20.
Graham SE, Clarke SL, Wu KH, et al: The power of genetic diversity in genome-wide association studies of lipids. Nature 2021; 600:675–679
21.
Evangelou E, Warren HR, Mosen-Ansorena D, et al: Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat Genet 2018; 50:1412–1425
22.
Andreassen OA, Djurovic S, Thompson WK, et al: Improved detection of common variants associated with schizophrenia by leveraging pleiotropy with cardiovascular-disease risk factors. Am J Hum Genet 2013; 92:197–209
23.
Hansen T, Ingason A, Djurovic S, et al: At-risk variant in TCF7L2 for type II diabetes increases risk of schizophrenia. Biol Psychiatry 2011; 70:59–63
24.
Hackinger S, Prins B, Mamakou V, et al: Evidence for genetic contribution to the increased risk of type 2 diabetes in schizophrenia. Transl Psychiatry 2018; 8:252
25.
Bahrami S, Steen NE, Shadrin A, et al: Shared genetic loci between body mass index and major psychiatric disorders: a genome-wide association study. JAMA Psychiatry 2020; 77:503–512
26.
Andreassen OA, McEvoy LK, Thompson WK, et al: Identifying common genetic variants in blood pressure due to polygenic pleiotropy with associated phenotypes. Hypertension 2014; 63:819–826
27.
Peters T, Nüllig L, Antel J, et al: The role of genetic variation of BMI, body composition, and fat distribution for mental traits and disorders: a look-up and Mendelian randomization study. Front Genet 2020; 11:373
28.
Rødevand L, Bahrami S, Frei O, et al: Extensive bidirectional genetic overlap between bipolar disorder and cardiovascular disease phenotypes. Transl Psychiatry 2021; 11:407
29.
Strawbridge RJ, Johnston KJA, Bailey MES, et al: The overlap of genetic susceptibility to schizophrenia and cardiometabolic disease can be used to identify metabolically different groups of individuals. Sci Rep 2021; 11:632
30.
Liu M, Jiang Y, Wedow R, et al: Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat Genet 2019; 51:237–244
31.
Peterson RE, Bigdeli TB, Ripke S, et al: Genome-wide analyses of smoking behaviors in schizophrenia: findings from the Psychiatric Genomics Consortium. J Psychiatr Res 2021; 137:215–224
32.
Smeland OB, Frei O, Shadrin A, et al: Discovery of shared genomic loci using the conditional false discovery rate approach. Hum Genet 2020; 139:85–94
33.
Frei O, Holland D, Smeland OB, et al: Bivariate causal mixture model quantifies polygenic overlap between complex traits beyond genetic correlation. Nat Commun 2019; 10:2417
34.
1000 Genomes Project Consortium, Auton A, Brooks LD, et al: A global reference for human genetic variation. Nature 2015; 526:68–74
35.
Lokki ML, Paakkanen R: The complexity and diversity of major histocompatibility complex challenge disease association studies. HLA 2019; 93:3–15
36.
Tiwari AK, Zai CC, Müller DJ, et al: Genetics in schizophrenia: where are we and what next? Dialogues Clin Neurosci 2010; 12:289–303
37.
Watanabe K, Taskesen E, van Bochoven A, et al: Functional mapping and annotation of genetic associations with FUMA. Nat Commun 2017; 8:1826
38.
Bulik-Sullivan BK, Loh PR, Finucane HK, et al: LD score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet 2015; 47:291–295
39.
Bahrami S, Hindley G, Winsvold BS, et al: Dissecting the shared genetic basis of migraine and mental disorders using novel statistical tools. Brain 2022; 145:142–153
40.
Wiström ED, O’Connell KS, Karadag N, et al: Genome-wide analysis reveals genetic overlap between alcohol use behaviours, schizophrenia and bipolar disorder and identifies novel shared risk loci. Addiction 2022; 117:600–610
41.
Lee JJ, Wedow R, Okbay A, et al: Gene discovery and polygenic prediction from a genome-wide association study of educational attainment in 1.1 million individuals. Nat Genet 2018; 50:1112–1121
42.
Savage JE, Jansen PR, Stringer S, et al: Genome-wide association meta-analysis in 269,867 individuals identifies new genetic and functional links to intelligence. Nat Genet 2018; 50:912–919
43.
Cheng W, Frei O, van der Meer D, et al: Genetic association between schizophrenia and cortical brain surface area and thickness. JAMA Psychiatry 2021; 78:1020–1030
44.
Hindley G, O’Connell KS, Rahman Z, et al: The shared genetic basis of mood instability and psychiatric disorders: a cross-trait genome-wide association analysis. Am J Med Genet B Neuropsychiatr Genet 2022; 189:207–218
45.
Akiyama M, Okada Y, Kanai M, et al: Genome-wide association study identifies 112 new loci for body mass index in the Japanese population. Nat Genet 2017; 49:1458–1467
46.
Giri A, Hellwege JN, Keaton JM, et al: Trans-ethnic association study of blood pressure determinants in over 750,000 individuals. Nat Genet 2019; 51:51–62
47.
Koyama S, Ito K, Terao C, et al: Population-specific and trans-ancestry genome-wide analyses identify distinct and shared genetic risk loci for coronary artery disease. Nat Genet 2020; 52:1169–1177
48.
Matoba N, Akiyama M, Ishigaki K, et al: GWAS of smoking behaviour in 165,436 Japanese people reveals seven new loci and shared genetic architecture. Nat Hum Behav 2019; 3:471–477
49.
Kircher M, Witten DM, Jain P, et al: A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014; 46:310–315
50.
Boyle AP, Hong EL, Hariharan M, et al: Annotation of functional variation in personal genomes using RegulomeDB. Genome Res 2012; 22:1790–1797
51.
Roadmap Epigenomics Consortium, Kundaje A, Meuleman W, et al: Integrative analysis of 111 reference human epigenomes. Nature 2015; 518:317–330
52.
Zhu Z, Zhang F, Hu H, et al: Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet 2016; 48:481–487
53.
Uffelmann E, Huang QQ, Munung NS, et al: Genome-wide association studies. Nat Rev Methods Primers 2021; 1:59
54.
Morris JA, Domingo J, Daniloski Z, et al: Discovery of target genes and pathways at GWAS loci by pooled single-cell CRISPR screens. Science 2023; 380:eadh7699
55.
Wormington B, Thorp JG, Scott JG, et al: Influences on the genetic relationship between cannabis use and schizophrenia: the role of the externalizing spectrum. Schizophr Bull 2022; 48:1318–1326
56.
Grotzinger AD, Rhemtulla M, de Vlaming R, et al: Genomic structural equation modelling provides insights into the multivariate genetic architecture of complex traits. Nat Hum Behav 2019; 3:513–525
57.
Holland D, Frei O, Desikan R, et al: Beyond SNP heritability: polygenicity and discoverability of phenotypes estimated with a univariate Gaussian mixture model. PLoS Genet 2020; 16:e1008612
58.
Inamura Y, Sagae T, Nakamachi K, et al: Body mass index of inpatients with schizophrenia in Japan. Int J Psychiatry Med 2012; 44:171–181
59.
Sørensen HJ, Mortensen EL, Reinisch JM, et al: Height, weight and body mass index in early adulthood and risk of schizophrenia. Acta Psychiatr Scand 2006; 114:49–54
60.
Ringen PA, Engh JA, Birkenaes AB, et al: Increased mortality in schizophrenia due to cardiovascular disease: a non-systematic review of epidemiology, possible causes, and interventions. Front Psychiatry 2014; 5:137
61.
Jakobsen AS, Speyer H, Nørgaard HCB, et al: Dietary patterns and physical activity in people with schizophrenia and increased waist circumference. Schizophr Res 2018; 199:109–115
62.
Gebhardt S, Theisen FM, Haberhausen M, et al: Body weight gain induced by atypical antipsychotics: an extension of the monozygotic twin and sib pair study. J Clin Pharm Ther 2010; 35:207–211
63.
Jee SH, Lee SY, Nam CM, et al: Effect of smoking on the paradox of high waist-to-hip ratio and low body mass index. Obes Res 2002; 10:891–895
64.
So HC, Chau KL, Ao FK, et al: Exploring shared genetic bases and causal relationships of schizophrenia and bipolar disorder with 28 cardiovascular and metabolic traits. Psychol Med 2019; 49:1286–1298
65.
Duncan LE, Shen H, Ballon JS, et al: Genetic correlation profile of schizophrenia mirrors epidemiological results and suggests link between polygenic and rare variant (22q11.2) cases of schizophrenia. Schizophr Bull 2018; 44:1350–1361
66.
Shah P, Iwata Y, Caravaggio F, et al: Alterations in body mass index and waist-to-hip ratio in never and minimally treated patients with psychosis: a systematic review and meta-analysis. Schizophr Res 2019; 208:420–429
67.
Caponnetto P, Polosa R, Robson D, et al: Tobacco smoking, related harm and motivation to quit smoking in people with schizophrenia spectrum disorders. Health Psychol Res 2020; 8:9042
68.
Parikh V, Kutlu MG, Gould TJ: nAChR dysfunction as a common substrate for schizophrenia and comorbid nicotine addiction: current trends and perspectives. Schizophr Res 2016; 171:1–15
69.
Quach BC, Bray MJ, Gaddis NC, et al: Expanding the genetic architecture of nicotine dependence and its shared genetics with multiple traits. Nat Commun 2020; 11:5562
70.
Harris LW, Guest PC, Wayland MT, et al: Schizophrenia: metabolic aspects of aetiology, diagnosis and future treatment strategies. Psychoneuroendocrinology 2013; 38:752–766
71.
Wu X, Huang Z, Wu R, et al: The comparison of glycometabolism parameters and lipid profiles between drug-naïve, first-episode schizophrenia patients and healthy controls. Schizophr Res 2013; 150:157–162
72.
Mistry S, Harrison JR, Smith DJ, et al: The use of polygenic risk scores to identify phenotypes associated with genetic risk of schizophrenia: systematic review. Schizophr Res 2018; 197:2–8
73.
Storch Jakobsen A, Speyer H, Nørgaard HCB, et al: Associations between clinical and psychosocial factors and metabolic and cardiovascular risk factors in overweight patients with schizophrenia spectrum disorders: baseline and two-years findings from the CHANGE trial. Schizophr Res 2018; 199:96–102
74.
Arango C, Bobes J, Kirkpatrick B, et al: Psychopathology, coronary heart disease and metabolic syndrome in schizophrenia spectrum patients with deficit versus non-deficit schizophrenia: findings from the CLAMORS study. Eur Neuropsychopharmacol 2011; 21:867–875
75.
Fernández-Ruiz I: Immune system and cardiovascular disease. Nat Rev Cardiol 2016; 13:503
Information & Authors
Information
Published In


History
Received: 1 August 2022
Revision received: 23 April 2023
Accepted: 26 May 2023
Published online: 27 September 2023
Published in print: November 1, 2023
Keywords
Authors
Funding Information
Supported by the Research Council of Norway (grants 223273, 300309, 324252, and 326813), the Norwegian Health Association (grant 22731), K.G. Jebsen Stiftelsen, the South-East Norway Regional Health Authority (grants 2019-108 and 2017-112), and the European Union’s Horizon 2020 Research and Innovation Action Grant (CoMorMent grant 847776).Dr. Dale is a founder of and holds equity interest in CorTechs Labs and serves on its scientific advisory board; he is a member of the scientific advisory board of Human Longevity and the Mohn Medical Imaging and Visualization Center (Bergen, Norway); and he has received research funding from General Electric Healthcare. Dr. Andreassen has served as a speaker for Janssen, Lundbeck, and Sunovion and as a consultant for Cortechs.ai. The other authors report no financial relationships with commercial interests.
Metrics & Citations
Metrics
Citations
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
For more information or tips please see 'Downloading to a citation manager' in the Help menu.
View Options
View options
PDF/EPUB
View PDF/EPUBFigures
FIGURE 1. Shared and unique polygenic components between schizophrenia and cardiovascular disease phenotypesa
aThe Venn diagrams illustrate shared and unique trait-influencing variants, showing polygenic overlap (green) between schizophrenia (blue) and cardiovascular disease phenotypes (orange), including A) smoking initiation, B) body mass index (BMI), C) systolic blood pressure (BP), D) diastolic BP, E) type 2 diabetes, and F) high-density lipoprotein (HDL). The numbers in the Venn diagram indicate the estimated quantity of shared and unique trait-influencing variants (in thousands), explaining 90% of single-nucleotide polymorphism heritability in each phenotype, and the standard error. The size of the circles reflects the degree of polygenicity. The genetic correlations (rg) are also provided. The figures are based on MiXeR analysis.
FIGURE 2. Common genetic variants jointly associated with schizophrenia and cardiovascular disease phenotypesa
aThe Manhattan plots show the common genetic variants jointly associated with schizophrenia and A) body mass index (BMI), B) waist-to-hip ratio (adjusted for BMI), C) smoking initiation, D) cigarettes per day, E) triglycerides, F) high-density lipoprotein (HDL), G) low-density lipoprotein (LDL), H) total cholesterol, I) systolic blood pressure (BP), J) diastolic BP, K) type 2 diabetes, and L) coronary artery disease at conjunctional false discovery rate (conjFDR)<0.05. Each panel shows the –log10 transformed conjFDR values for each single-nucleotide polymorphism (SNP) on the y-axis and chromosomal positions along the x-axis. SNPs with conjFDR<0.05 (i.e., −log10 FDR>1.3) are shown with enlarged data points. A black circle around the enlarged data points indicates the most significant SNP in each linkage disequilibrium block. The figure shows the localization of the “conjunctional loci”; additional details are provided in the tables in the online supplement .
Tables
Media
References
References
1.
Heiberg IH, Jacobsen BK, Nesvåg R, et al: Total and cause-specific standardized mortality ratios in patients with schizophrenia and/or substance use disorder. PLoS One 2018; 13:e0202028
2.
Laursen TM, Plana-Ripoll O, Andersen PK, et al: Cause-specific life years lost among persons diagnosed with schizophrenia: is it getting better or worse? Schizophr Res 2019; 206:284–290
3.
Rødevand L, Steen NE, Elvsåshagen T, et al: Cardiovascular risk remains high in schizophrenia with modest improvements in bipolar disorder during past decade. Acta Psychiatr Scand 2019; 139:348–360
4.
Plana-Ripoll O, Weye N, Momen NC, et al: Changes over time in the differential mortality gap in individuals with mental disorders. JAMA Psychiatry 2020; 77:648–650
5.
Lambert AM, Parretti HM, Pearce E, et al: Temporal trends in associations between severe mental illness and risk of cardiovascular disease: a systematic review and meta-analysis. PLoS Med 2022; 19:e1003960
6.
Correll CU, Detraux J, De Lepeleire J, et al: Effects of antipsychotics, antidepressants and mood stabilizers on risk for physical diseases in people with schizophrenia, depression and bipolar disorder. World Psychiatry 2015; 14:119–136
7.
Badcock JC, Mackinnon A, Waterreus A, et al: Loneliness in psychotic illness and its association with cardiometabolic disorders. Schizophr Res 2019; 204:90–95
8.
De Hert M, Correll CU, Bobes J, et al: Physical illness in patients with severe mental disorders. I. prevalence, impact of medications and disparities in health care. World Psychiatry 2011; 10:52–77
9.
Mothi SS, Tandon N, Padmanabhan J, et al: Increased cardiometabolic dysfunction in first-degree relatives of patients with psychotic disorders. Schizophr Res 2015; 165:103–107
10.
Yang W, Zheng L, Zheng B, et al: A meta-analysis of abnormal glucose metabolism in first-episode drug-naive schizophrenia. Psychiatr Danub 2020; 32:46–54
11.
Zammit S, Rasmussen F, Farahmand B, et al: Height and body mass index in young adulthood and risk of schizophrenia: a longitudinal study of 1 347 520 Swedish men. Acta Psychiatr Scand 2007; 116:378–385
12.
Pérez-Piñar M, Mathur R, Foguet Q, et al: Cardiovascular risk factors among patients with schizophrenia, bipolar, depressive, anxiety, and personality disorders. Eur Psychiatry 2016; 35:8–15
13.
Sullivan PF, Kendler KS, Neale MC: Schizophrenia as a complex trait: evidence from a meta-analysis of twin studies. Arch Gen Psychiatry 2003; 60:1187–1192
14.
Smeland OB, Frei O, Dale AM, et al: The polygenic architecture of schizophrenia: rethinking pathogenesis and nosology. Nat Rev Neurol 2020; 16:366–379
15.
Trubetskoy V, Pardiñas AF, Qi T, et al: Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature 2022; 604:502–508
16.
Nelson CP, Goel A, Butterworth AS, et al: Association analyses based on false discovery rate implicate new loci for coronary artery disease. Nat Genet 2017; 49:1385–1391
17.
Turcot V, Lu Y, Highland HM, et al: Protein-altering variants associated with body mass index implicate pathways that control energy intake and expenditure in obesity. Nat Genet 2018; 50:26–41
18.
Pulit SL, Stoneman C, Morris AP, et al: Meta-analysis of genome-wide association studies for body fat distribution in 694 649 individuals of European ancestry. Hum Mol Genet 2019; 28:166–174
19.
Mahajan A, Taliun D, Thurner M, et al: Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat Genet 2018; 50:1505–1513
20.
Graham SE, Clarke SL, Wu KH, et al: The power of genetic diversity in genome-wide association studies of lipids. Nature 2021; 600:675–679
21.
Evangelou E, Warren HR, Mosen-Ansorena D, et al: Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat Genet 2018; 50:1412–1425
22.
Andreassen OA, Djurovic S, Thompson WK, et al: Improved detection of common variants associated with schizophrenia by leveraging pleiotropy with cardiovascular-disease risk factors. Am J Hum Genet 2013; 92:197–209
23.
Hansen T, Ingason A, Djurovic S, et al: At-risk variant in TCF7L2 for type II diabetes increases risk of schizophrenia. Biol Psychiatry 2011; 70:59–63
24.
Hackinger S, Prins B, Mamakou V, et al: Evidence for genetic contribution to the increased risk of type 2 diabetes in schizophrenia. Transl Psychiatry 2018; 8:252
25.
Bahrami S, Steen NE, Shadrin A, et al: Shared genetic loci between body mass index and major psychiatric disorders: a genome-wide association study. JAMA Psychiatry 2020; 77:503–512
26.
Andreassen OA, McEvoy LK, Thompson WK, et al: Identifying common genetic variants in blood pressure due to polygenic pleiotropy with associated phenotypes. Hypertension 2014; 63:819–826
27.
Peters T, Nüllig L, Antel J, et al: The role of genetic variation of BMI, body composition, and fat distribution for mental traits and disorders: a look-up and Mendelian randomization study. Front Genet 2020; 11:373
28.
Rødevand L, Bahrami S, Frei O, et al: Extensive bidirectional genetic overlap between bipolar disorder and cardiovascular disease phenotypes. Transl Psychiatry 2021; 11:407
29.
Strawbridge RJ, Johnston KJA, Bailey MES, et al: The overlap of genetic susceptibility to schizophrenia and cardiometabolic disease can be used to identify metabolically different groups of individuals. Sci Rep 2021; 11:632
30.
Liu M, Jiang Y, Wedow R, et al: Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat Genet 2019; 51:237–244
31.
Peterson RE, Bigdeli TB, Ripke S, et al: Genome-wide analyses of smoking behaviors in schizophrenia: findings from the Psychiatric Genomics Consortium. J Psychiatr Res 2021; 137:215–224
32.
Smeland OB, Frei O, Shadrin A, et al: Discovery of shared genomic loci using the conditional false discovery rate approach. Hum Genet 2020; 139:85–94
33.
Frei O, Holland D, Smeland OB, et al: Bivariate causal mixture model quantifies polygenic overlap between complex traits beyond genetic correlation. Nat Commun 2019; 10:2417
34.
1000 Genomes Project Consortium, Auton A, Brooks LD, et al: A global reference for human genetic variation. Nature 2015; 526:68–74
35.
Lokki ML, Paakkanen R: The complexity and diversity of major histocompatibility complex challenge disease association studies. HLA 2019; 93:3–15
36.
Tiwari AK, Zai CC, Müller DJ, et al: Genetics in schizophrenia: where are we and what next? Dialogues Clin Neurosci 2010; 12:289–303
37.
Watanabe K, Taskesen E, van Bochoven A, et al: Functional mapping and annotation of genetic associations with FUMA. Nat Commun 2017; 8:1826
38.
Bulik-Sullivan BK, Loh PR, Finucane HK, et al: LD score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet 2015; 47:291–295
39.
Bahrami S, Hindley G, Winsvold BS, et al: Dissecting the shared genetic basis of migraine and mental disorders using novel statistical tools. Brain 2022; 145:142–153
40.
Wiström ED, O’Connell KS, Karadag N, et al: Genome-wide analysis reveals genetic overlap between alcohol use behaviours, schizophrenia and bipolar disorder and identifies novel shared risk loci. Addiction 2022; 117:600–610
41.
Lee JJ, Wedow R, Okbay A, et al: Gene discovery and polygenic prediction from a genome-wide association study of educational attainment in 1.1 million individuals. Nat Genet 2018; 50:1112–1121
42.
Savage JE, Jansen PR, Stringer S, et al: Genome-wide association meta-analysis in 269,867 individuals identifies new genetic and functional links to intelligence. Nat Genet 2018; 50:912–919
43.
Cheng W, Frei O, van der Meer D, et al: Genetic association between schizophrenia and cortical brain surface area and thickness. JAMA Psychiatry 2021; 78:1020–1030
44.
Hindley G, O’Connell KS, Rahman Z, et al: The shared genetic basis of mood instability and psychiatric disorders: a cross-trait genome-wide association analysis. Am J Med Genet B Neuropsychiatr Genet 2022; 189:207–218
45.
Akiyama M, Okada Y, Kanai M, et al: Genome-wide association study identifies 112 new loci for body mass index in the Japanese population. Nat Genet 2017; 49:1458–1467
46.
Giri A, Hellwege JN, Keaton JM, et al: Trans-ethnic association study of blood pressure determinants in over 750,000 individuals. Nat Genet 2019; 51:51–62
47.
Koyama S, Ito K, Terao C, et al: Population-specific and trans-ancestry genome-wide analyses identify distinct and shared genetic risk loci for coronary artery disease. Nat Genet 2020; 52:1169–1177
48.
Matoba N, Akiyama M, Ishigaki K, et al: GWAS of smoking behaviour in 165,436 Japanese people reveals seven new loci and shared genetic architecture. Nat Hum Behav 2019; 3:471–477
49.
Kircher M, Witten DM, Jain P, et al: A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014; 46:310–315
50.
Boyle AP, Hong EL, Hariharan M, et al: Annotation of functional variation in personal genomes using RegulomeDB. Genome Res 2012; 22:1790–1797
51.
Roadmap Epigenomics Consortium, Kundaje A, Meuleman W, et al: Integrative analysis of 111 reference human epigenomes. Nature 2015; 518:317–330
52.
Zhu Z, Zhang F, Hu H, et al: Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet 2016; 48:481–487
53.
Uffelmann E, Huang QQ, Munung NS, et al: Genome-wide association studies. Nat Rev Methods Primers 2021; 1:59
54.
Morris JA, Domingo J, Daniloski Z, et al: Discovery of target genes and pathways at GWAS loci by pooled single-cell CRISPR screens. Science 2023; 380:eadh7699
55.
Wormington B, Thorp JG, Scott JG, et al: Influences on the genetic relationship between cannabis use and schizophrenia: the role of the externalizing spectrum. Schizophr Bull 2022; 48:1318–1326
56.
Grotzinger AD, Rhemtulla M, de Vlaming R, et al: Genomic structural equation modelling provides insights into the multivariate genetic architecture of complex traits. Nat Hum Behav 2019; 3:513–525
57.
Holland D, Frei O, Desikan R, et al: Beyond SNP heritability: polygenicity and discoverability of phenotypes estimated with a univariate Gaussian mixture model. PLoS Genet 2020; 16:e1008612
58.
Inamura Y, Sagae T, Nakamachi K, et al: Body mass index of inpatients with schizophrenia in Japan. Int J Psychiatry Med 2012; 44:171–181
59.
Sørensen HJ, Mortensen EL, Reinisch JM, et al: Height, weight and body mass index in early adulthood and risk of schizophrenia. Acta Psychiatr Scand 2006; 114:49–54
60.
Ringen PA, Engh JA, Birkenaes AB, et al: Increased mortality in schizophrenia due to cardiovascular disease: a non-systematic review of epidemiology, possible causes, and interventions. Front Psychiatry 2014; 5:137
61.
Jakobsen AS, Speyer H, Nørgaard HCB, et al: Dietary patterns and physical activity in people with schizophrenia and increased waist circumference. Schizophr Res 2018; 199:109–115
62.
Gebhardt S, Theisen FM, Haberhausen M, et al: Body weight gain induced by atypical antipsychotics: an extension of the monozygotic twin and sib pair study. J Clin Pharm Ther 2010; 35:207–211
63.
Jee SH, Lee SY, Nam CM, et al: Effect of smoking on the paradox of high waist-to-hip ratio and low body mass index. Obes Res 2002; 10:891–895
64.
So HC, Chau KL, Ao FK, et al: Exploring shared genetic bases and causal relationships of schizophrenia and bipolar disorder with 28 cardiovascular and metabolic traits. Psychol Med 2019; 49:1286–1298
65.
Duncan LE, Shen H, Ballon JS, et al: Genetic correlation profile of schizophrenia mirrors epidemiological results and suggests link between polygenic and rare variant (22q11.2) cases of schizophrenia. Schizophr Bull 2018; 44:1350–1361
66.
Shah P, Iwata Y, Caravaggio F, et al: Alterations in body mass index and waist-to-hip ratio in never and minimally treated patients with psychosis: a systematic review and meta-analysis. Schizophr Res 2019; 208:420–429
67.
Caponnetto P, Polosa R, Robson D, et al: Tobacco smoking, related harm and motivation to quit smoking in people with schizophrenia spectrum disorders. Health Psychol Res 2020; 8:9042
68.
Parikh V, Kutlu MG, Gould TJ: nAChR dysfunction as a common substrate for schizophrenia and comorbid nicotine addiction: current trends and perspectives. Schizophr Res 2016; 171:1–15
69.
Quach BC, Bray MJ, Gaddis NC, et al: Expanding the genetic architecture of nicotine dependence and its shared genetics with multiple traits. Nat Commun 2020; 11:5562
70.
Harris LW, Guest PC, Wayland MT, et al: Schizophrenia: metabolic aspects of aetiology, diagnosis and future treatment strategies. Psychoneuroendocrinology 2013; 38:752–766
71.
Wu X, Huang Z, Wu R, et al: The comparison of glycometabolism parameters and lipid profiles between drug-naïve, first-episode schizophrenia patients and healthy controls. Schizophr Res 2013; 150:157–162
72.
Mistry S, Harrison JR, Smith DJ, et al: The use of polygenic risk scores to identify phenotypes associated with genetic risk of schizophrenia: systematic review. Schizophr Res 2018; 197:2–8
73.
Storch Jakobsen A, Speyer H, Nørgaard HCB, et al: Associations between clinical and psychosocial factors and metabolic and cardiovascular risk factors in overweight patients with schizophrenia spectrum disorders: baseline and two-years findings from the CHANGE trial. Schizophr Res 2018; 199:96–102
74.
Arango C, Bobes J, Kirkpatrick B, et al: Psychopathology, coronary heart disease and metabolic syndrome in schizophrenia spectrum patients with deficit versus non-deficit schizophrenia: findings from the CLAMORS study. Eur Neuropsychopharmacol 2011; 21:867–875
75.
Fernández-Ruiz I: Immune system and cardiovascular disease. Nat Rev Cardiol 2016; 13:503