The Psychiatric Presentation of Mitochondrial Disorders in Adults
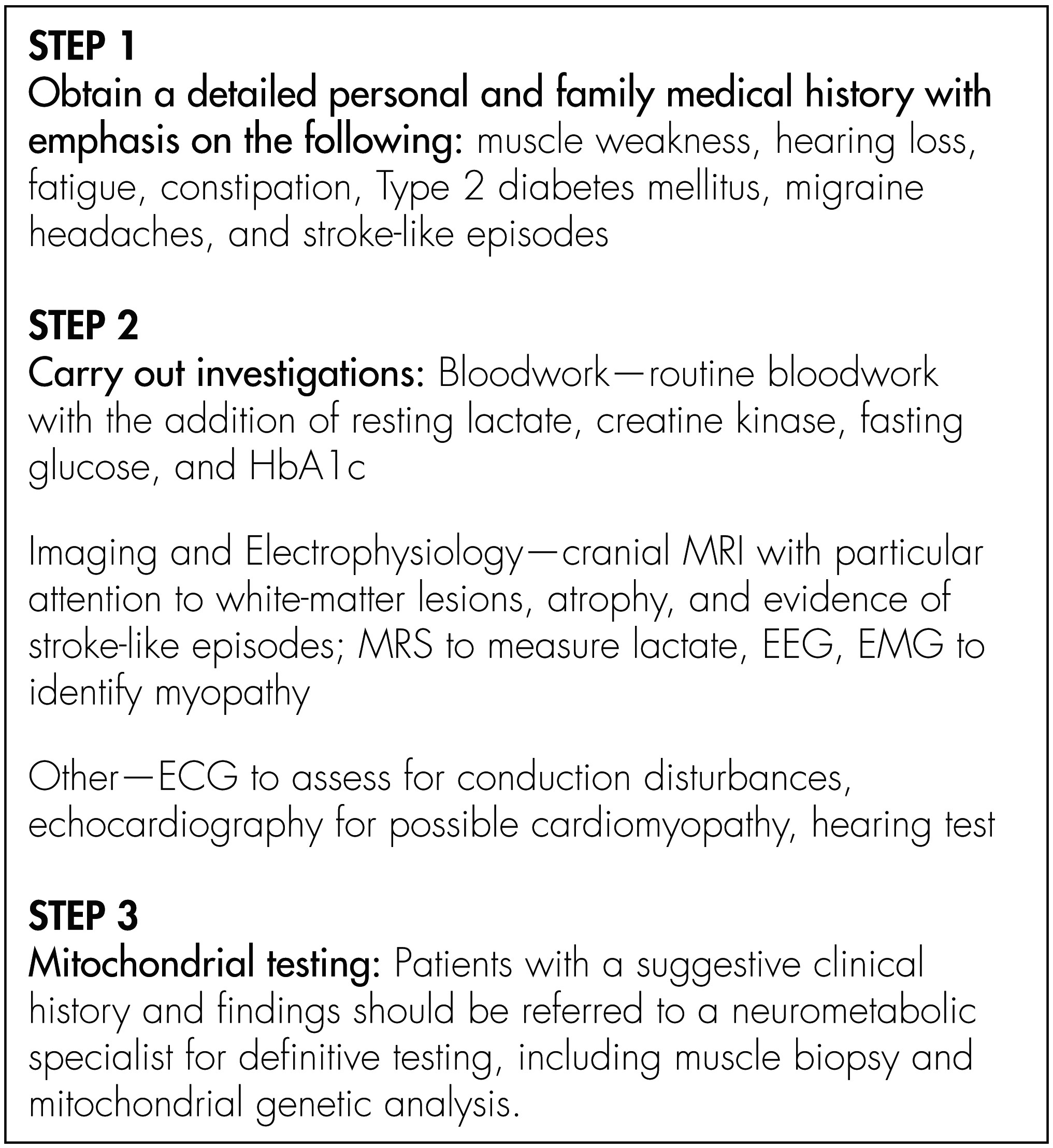
Method
Case Series
Review of the Literature
Results
Cases of Major Depressive Disorder
Case 1:
Case 2:
Case 3:
Case 4:
Case 5:
Case 6:
Case 7:
Case of Anorexia Nervosa
Case 8:
Cases of Bipolar Affective Disorder With Atypical Features
Case 9:
Case 10:
Cases of Treatment-Resistant Obsessive-Compulsive Disorder (OCD)
Case 11:
Case 12:
Review of the Literature
| Case | Sex | Onset of Psychiatric Symptoms, years | Age at Diagnosis, years | Major Depressive Disorder | Bipolar Affective Disorder | Psychosis | OCD or Anxiety | Cognitive Deterioration | Frontal Lobe Syndrome | Personality Change | Diagnosis | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F | 16 | NR | + | — | + | — | — | — | — | Mitochondrial cytopathy | Ahn et al., 200514 |
| 2 | M | 29 | 36 | — | — | + | — | + | — | — | C3256T | Amemiya et al., 200015 |
| 3 | F | 58 | 58 | — | — | + | — | + | — | — | MELAS 3243 | Apostolova et al., 200516 |
| 4 | M | 25 | 37 | — | — | + | — | — | — | — | MELAS 3243 | Ban et al., 199217 |
| 5 | F | NR | 28 | + | — | + | — | — | — | — | MELAS | Bhuvaneswar et al., 200856 |
| 6 | F | 44 | 44 | — | — | + | — | + | — | — | MELAS 3243 | Clark et al., 199618 |
| 7 | F | NR | 33 | + | — | + | — | — | — | — | C3303T | Campos et al., 200119 |
| 8 | M | 24 | 46 | — | — | + | — | — | — | — | KSS | Desnuelle et al., 198920 |
| 9 | M | 22 | 61 | + | — | + | — | — | — | — | Mitochondrial cytopathy | Gardner et al., 200321 |
| 10 | M | 22 | 61 | + | — | + | — | — | — | — | Mitochondrial cytopathy | Grover et al., 200646 |
| 11 | F | 16 | NR | + | — | + | — | — | — | — | POLG1 mutation | Hopkins et al., 201047 |
| 12 | F | 17 | NR | + | — | + | — | — | — | — | POLG1 mutation | Hopkins et al., 201047 |
| 13 | M | 31 | 35 | — | — | + | — | + | + | — | MELAS 3243 | Inagaki et al., 199722 |
| 14 | M | 27 | 33 | + | — | + | — | + | — | — | MELAS 3274 | Jaksch et al., 200123 |
| 15 | F | 53 | 53 | — | — | + | — | — | — | — | MELAS 3243 | Kaido et al., 199624 |
| 16 | M | 32 | 32 | — | — | + | — | + | — | — | MELAS | Kiejna et al., 200225 |
| 17 | F | 12 | 16 | + | — | + | — | + | — | — | PDHA1 mutation | Koene et al., 200948 |
| 18 | F | 14 | 16 | + | — | — | — | — | — | — | MTND1 mutation | Koene et al., 200948 |
| 19 | F | 16 | 17 | + | — | + | — | — | — | — | POLG1 mutation | Koene et al., 200948 |
| 20 | F | 15 | 16 | + | — | — | — | + | — | — | MTTK mutation | Koene et al., 200948 |
| 21 | M | 34 | 37 | — | — | — | — | — | + | + | MELAS 3243 | Koller et al., 200326 |
| 22 | F | NR | NR | + | — | + | — | + | — | — | POLG mutation | Komulainen et al., 201049 |
| 23 | M | 30 | 30 | — | — | — | + | — | — | — | MELAS 3243 | Lacey et al., 200827 |
| 24 | M | 49 | 51 | — | — | — | + | — | — | — | MELAS 3243 | Lacey et al., 200827 |
| 25 | F | 12 | 63 | + | — | — | — | + | — | — | Mt DNA deletions | Mancuso et al,. 200850 |
| 26 | M | 47 | 52 | + | — | — | — | — | — | MELAS 3243 | Miyaoka et al., 199728 | |
| 27 | F | 37 | 62 | — | — | — | + | — | — | — | MELAS 3243 | Miyaoka et al., 199728 |
| 28 | F | 25 | 69 | — | — | — | + | — | — | — | MELAS 3243 | Miyaoka et al., 199728 |
| 29 | M | 35 | 61 | — | — | — | + | — | — | — | MELAS 3243 | Miyaoka et al., 199728 |
| 30 | F | 23 | 52 | — | — | + | — | + | — | — | MELAS | Odawara et al., 199731 |
| 31 | M | 22 | 23 | + | — | — | + | — | — | — | MELAS 3243 | Onishi et al., 199732 |
| 32 | F | 27 | 47 | — | — | + | — | — | — | — | MELAS 3243 | Prayson et al., 199833 |
| 33 | M | 30 | 47 | + | — | — | — | + | — | — | POLG mutation | Rantamaki et al., 200152 |
| 34 | M | 28 | 34 | — | — | + | — | — | + | + | MELAS 3243 | Sartor et al., 200235 |
| 35 | F | 48 | 48 | — | — | + | — | + | — | — | MELAS 3243 | Shinkai et al., 200037 |
| 36 | F | NR | 44 | — | + | + | — | — | — | — | ANT1 mutation | Siciliano et al., 200353 |
| 37 | M | 21 | 36 | — | — | + | — | — | — | MELAS 3243 | Spellberg et al., 200138 | |
| 38 | F | 23 | 44 | — | + | — | — | + | — | — | KSS | Stewart and Naylor, 199039 |
| 39 | F | 19 | 29 | + | — | + | — | + | — | AD PEO, Twinkle mutation | Suomalainen et al., 199240 | |
| 40 | M | 25 | 35 | — | — | + | — | + | — | — | MELAS | Suzuki et al., 198941 |
| 41 | F | 29 | NR | + | — | + | — | — | — | — | MELAS 3251 | Sweeney et al., 199343 |
| 42 | F | 25 | 39 | — | — | — | — | — | — | — | MELAS 3251 | Sweeney et al., 199343 |
| 43 | F | 22 | 27 | — | — | + | — | + | — | — | MELAS 3243 | Thomeer et al., 199844 |
| 44 | M | 23 | 39 | NR | NR | NR | NR | NR | NR | NR | POLG mutation | Van Goethem et al., 200451 |
| 45 | F | 37 | 52 | + | — | + | — | — | — | — | POLG mutation | Verhoeven et al., 201154 |
| 46 | M | 29 | 37 | — | — | + | — | + | — | — | MELAS | Yamazaki et al., 199145 |
| 47 | F | 57 | NR | + | — | + | — | + | — | — | MTTF mutation | Young et al., 201055 |
| Mean/Total | 29 | 41 | 20 | 2 | 30 | 6 | 18 | 4 | 2 | |||
Discussion
Psychiatric Features
| Patient/Sex | 1/F | 2/F | 3/M | 4/M | 5/M | 6/F | 7/M | 8/F | 9/F | 10/F | 11/M | 12/F | Mean Total |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age at onset psychiatric disorder, years | 53 | 47 | 41 | 48 | 38 | 16 | 30 | 20 | 20 | 17 | 15 | 42 | 32 |
| Age at diagnosis of mitochondrial disorder, years | 54 | 50 | 54 | 52 | 43 | 45 | 46 | 24 | 46 | 54 | 24 | 48 | 45 |
| Family psychiatric history | + | — | + | — | — | + | + | — | + | — | — | + | 6 |
| Treatment-resistant illness | + | + | + | + | + | + | — | + | + | + | + | + | 11 |
| Deterioration on psychotropic medications | — | — | — | + | — | + | — | — | + | — | — | — | 3 |
| Mood disorder | 12 | ||||||||||||
| Major depressive disorder | + | + | + | + | + | + | + | + | — | — | + | + | 10 |
| With psychotic features | — | — | — | + | + | + | — | — | — | — | — | — | 3 |
| Bipolar Affective disorder | — | — | — | — | — | — | — | — | + | + | — | — | 2 |
| With psychotic features | — | — | — | — | — | — | — | — | — | + | — | — | 1 |
| Anxiety disorder | 7 | ||||||||||||
| Generalized anxiety disorder | — | + | + | — | — | — | + | — | — | — | + | + | 5 |
| Social anxiety | + | — | — | — | — | — | — | — | — | — | + | + | 3 |
| Obsessive-compulsive disorder | — | + | — | — | — | — | — | — | — | — | + | + | 3 |
| Panic disorder | — | — | + | — | — | — | — | — | + | — | — | — | 2 |
| Cognitive disorder | 6 | ||||||||||||
| Cognitive deterioration | + | + | — | + | — | + | — | — | — | + | — | + | 6 |
| Frontal lobe syndrome | — | — | — | + | — | — | — | — | — | + | — | + | 3 |
| Other | |||||||||||||
| Anorexia nervosa | — | — | — | — | — | — | — | + | — | — | — | + | 2 |
| Borderline personality disorder | — | — | — | — | — | + | — | — | — | — | — | — | 1 |
| Catatonia | — | — | — | + | — | — | — | — | — | — | — | — | 1 |
| Substance abuse or dependence | — | — | — | — | — | — | + | — | — | — | — | — | 1 |
Physical Features and Past Medical History
| Patient/Sex | 1/F | 2/F | 3/M | 4/M | 5/M | 6/F | 7/M | 8/F | 9/F | 10/F | 11/M | 12/F | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mitochondrial disorder diagnosis | MELAS 3243 | MELAS 3271 | Novel mutation: 11081A>T | 3.3kb deletion | CPEO 7.5kb deletion | MELAS 3271 | Novel mutation: 760A>G | MNGIE | MERRF 8363G>A | Unknown | MERRF 8344 | MELAS 3243 | Total |
| Medical Histories | |||||||||||||
| Muscle weakness or atrophy | + | — | — | + | + | + | + | + | + | + | + | + | 10 |
| Hearing loss | + | + | — | + | — | + | + | + | + | — | — | + | 8 |
| Fatigue | + | + | — | + | + | + | + | + | + | — | — | — | 8 |
| Dysphagia | — | — | — | + | + | — | + | + | + | — | + | — | 6 |
| Constipation | + | + | — | — | — | — | + | + | — | — | — | + | 5 |
| Stroke/stroke-like episodes | + | + | — | — | — | + | + | — | — | + | — | — | 5 |
| Type 2 diabetes | + | — | — | — | — | + | — | — | + | — | — | + | 4 |
| Migraines or headaches | + | — | — | — | — | — | + | + | — | + | — | — | 4 |
| Cardiac conduction abnormality | — | — | + | + | — | — | — | — | — | + | — | — | 3 |
| Short stature | — | — | — | — | — | + | — | — | — | + | — | + | 3 |
| Dysarthria | — | — | — | + | — | — | — | — | — | + | + | — | 3 |
| Cardiomyopathy | — | + | + | — | — | — | + | — | — | — | — | — | 3 |
| Seizure disorder | — | — | — | — | — | — | + | + | — | + | — | — | 3 |
| Lipomas | — | — | — | — | — | — | — | — | + | — | + | — | 2 |
| Dystonia | — | — | + | — | — | — | — | — | — | — | + | — | 2 |
| Arrhythmia | — | + | — | — | — | — | — | — | — | — | — | — | 1 |
| Recurrent miscarriages | — | — | — | — | — | — | — | — | — | — | — | + | 1 |
| Sleep apnea | — | — | + | — | — | — | — | — | — | — | — | — | 1 |
| Premature ovarian failure | — | — | — | — | — | — | — | — | + | — | — | — | 1 |
| Physical Examination | |||||||||||||
| Ataxia | + | — | — | + | + | + | — | — | + | + | + | — | 7 |
| Impaired vibration sense | + | — | — | + | — | — | + | — | + | + | + | + | 7 |
| Ptosis | + | — | — | + | + | — | + | + | — | — | — | + | 6 |
| Ophthalmoplegia | — | — | — | + | + | — | + | + | — | — | — | + | 5 |
| Babinski sign | — | — | — | + | — | + | — | — | — | + | + | — | 4 |
| Positive Romberg sign | — | — | — | + | — | — | — | — | + | + | — | — | 3 |
| Retinitis pigmentosa | + | — | — | + | — | — | — | — | — | — | — | — | 2 |
| Optic atrophy | + | — | — | — | — | — | — | — | — | — | + | — | 2 |
| Parkinsonism | + | — | + | — | — | — | — | — | — | — | — | — | 2 |
| Cataracts | — | + | + | — | — | — | + | — | — | — | — | — | 3 |
| Myoclonus or dyskinesia | — | — | — | — | + | — | + | — | — | + | — | — | 3 |
| Nystagmus | — | — | — | — | — | — | — | — | + | — | — | + | 2 |
| Hyperreflexia | — | — | — | — | — | + | — | — | — | + | — | — | 2 |
Laboratory Investigations
Neuroradiologic Findings
| Patient/Sex | 1/F | 2/F | 3/M | 4/M | 5/M | 6/F | 7/M | 8/F | 9/F | 10/F | 11/M | 12/F | Total/Mean |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Laboratory Investigations | |||||||||||||
| Elevated lactate | + | + | — | + | + | + | + | — | + | + | + | + | 10/12 |
| Maximum lactate (N: 0.7–2.2 mmol/L) | 4.4 | 6.8 | — | 7.9 | 6.7 | 4.7 | 4.9 | — | 2.5 | 4.4 | 6.1 | 2.7 | 5.1 |
| Elevated ammonia | + | — | — | + | + | — | — | — | — | — | — | — | 3/12 |
| Low pyruvate | — | — | — | — | — | — | — | — | — | + | — | + | 2/12 |
| Neuroimaging | |||||||||||||
| MRI: | |||||||||||||
| Normal | — | — | + | — | — | — | — | — | — | — | X | — | 1/11 |
| Atrophy | + | — | — | + | + | — | — | — | — | + | X | — | 4/11 |
| White-matter lesions | + | + | — | + | + | + | + | + | + | + | X | + | 10/11 |
| Lactate on MRS | — | + | — | — | + | + | — | — | — | + | X | — | 4/11 |
| CT: | |||||||||||||
| Normal | — | X | X | X | X | X | X | X | X | X | + | X | 1/2 |
| Atrophy | — | X | X | X | X | X | X | X | X | X | — | X | 0/2 |
| Basal ganglia calcifications | + | X | X | X | X | X | X | X | X | X | — | X | 1/2 |
| Electrodiagnostics | |||||||||||||
| EEG | A | A | X | X | X | N | X | A | X | A | X | N | 4A/6 |
| Auditory EVP | A | A | X | X | N | A | X | X | A | A | X | A | 6A/7 |
| Visual EVP | A | A | X | X | A | A | X | X | N | N | A | A | 6A/8 |
| Neuropsychiatric testing | X | X | N | A | X | A | X | X | A | A | X | A | 5A/6 |
| Muscle biopsy | |||||||||||||
| Ragged red fibers | + | + | — | + | + | + | + | X | — | — | X | + | 7/10 |
| Cox-negative fibers | — | — | — | + | + | — | — | X | + | — | X | + | 4/10 |
| Respiratory chain complex function | N | N | N | X | X | N | N | X | A | N | X | A | 2A/8 |
Electrophysiological Investigations
Muscle Biopsies
Correlation Between Clinical Phenotype and Genotype
Diagnostic and Treatment Considerations
Course of Illness
Limitations
Supplementary Material
- View/Download
- 102.44 KB
References
Information & Authors
Information
Published In


History
Authors
Funding Information
Metrics & Citations
Metrics
Citations
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
For more information or tips please see 'Downloading to a citation manager' in the Help menu.
View Options
View options
PDF/EPUB
View PDF/EPUBLogin options
Already a subscriber? Access your subscription through your login credentials or your institution for full access to this article.
Personal login Institutional Login Open Athens loginNot a subscriber?
PsychiatryOnline subscription options offer access to the DSM-5-TR® library, books, journals, CME, and patient resources. This all-in-one virtual library provides psychiatrists and mental health professionals with key resources for diagnosis, treatment, research, and professional development.
Need more help? PsychiatryOnline Customer Service may be reached by emailing [email protected] or by calling 800-368-5777 (in the U.S.) or 703-907-7322 (outside the U.S.).