Approximately one-third of symptoms reported by patients in primary care and subspecialty settings remain medically unexplained after a complete evaluation.
1 The term somatization refers to patients with medically unexplained symptoms that seek medical attention because of suspected disease or individuals reporting symptoms out-of-proportion to their medical condition. Their symptoms commonly include abdominal pain, bloating, dizziness, chest pain, breathlessness, pelvic pain, food intolerance, palpitations, and back pain among others.
2 When considering symptoms that have “bothered [the patient] a lot” in the past month, 16%−33% have no demonstrable medical basis.
3 Patients with medically unexplained symptoms engage in a disproportionately high rate of medical care utilization, including outpatient visits, hospitalizations, and overall healthcare costs (averaging 4700 USD annually per individual).
4The somatosensory amplification construct has been theorized by Arthur Barsky and colleagues to serve a critical role in the pathophysiology of somatization.
5 Somatosensory amplification refers to the tendency to experience a wide range of benign bodily sensations as intrusive, intense, noxious, and disruptive. Several elements are associated with amplification including: 1) a heightened attentional focus on bodily sensations; 2) the tendency to select out certain relatively weak and infrequent sensations; 3) the disposition to react to these sensations with affect and cognitions that intensify them and make them more alarming and distressing.
5 Somatosensory amplification has also been linked to alexithymia,
6,7 and somatization has been associated with dysphoric-anxious mood and cognitive distortions, particularly pain catastrophizing.
8Neuroimaging techniques applied to somatoform disorder populations and related cognitive-affective neuroscience studies in healthy populations allow for the in vivo detection of neural circuit abnormalities associated with somatization generally, and with somatosensory amplification more specifically. In this perspective article, central nervous system convergence of visceral and somatic processing is first summarized. Thereafter, neuroimaging studies in somatization disorder, undifferentiated somatization disorder, and somatoform pain disorder are reviewed [functional neurological symptom disorder (i.e., conversion disorder) was previously discussed elsewhere
9]. Hypothesized cognitive amplifiers of visceral-somatic processing including negative expectation, negative attentional bias, and pain catastrophizing, along with affective modifiers including alexithymia and dysphoric-anxious mood are discussed by integrating neuroimaging findings in healthy populations. Lastly, a neurocircuit model of somatosensory amplification is suggested. We propose that somatosensory amplification involves abnormal interactions among large-scale neural systems mediating visceral-somatic perception, emotional processing/awareness, and cognitive control. Frontolimbic, subcortical, and brainstem structures are particularly linked to the neurocircuitry of abnormal symptom amplification.
The purpose of this article is to provide a theoretical neurocircuit framework through which physician-scientists may begin to understand brain-behavior relationships in somatosensory amplification, rather than to provide a comprehensive review of published studies on somatosensory amplification and somatoform disorders. Articles using functional magnetic resonance imaging (fMRI), single photon-emission computed tomography (SPECT) and positron emission tomography (PET) to investigate somatization disorder, undifferentiated somatoform disorder, and somatoform pain disorder were emphasized. Studies exploring other “functional” disorders including fibromyalgia, chronic fatigue syndrome, and irritable bowel syndrome were largely omitted to limit the discussion to DSM-IV somatoform disorders.
Somatic and Visceral Afferent Processing
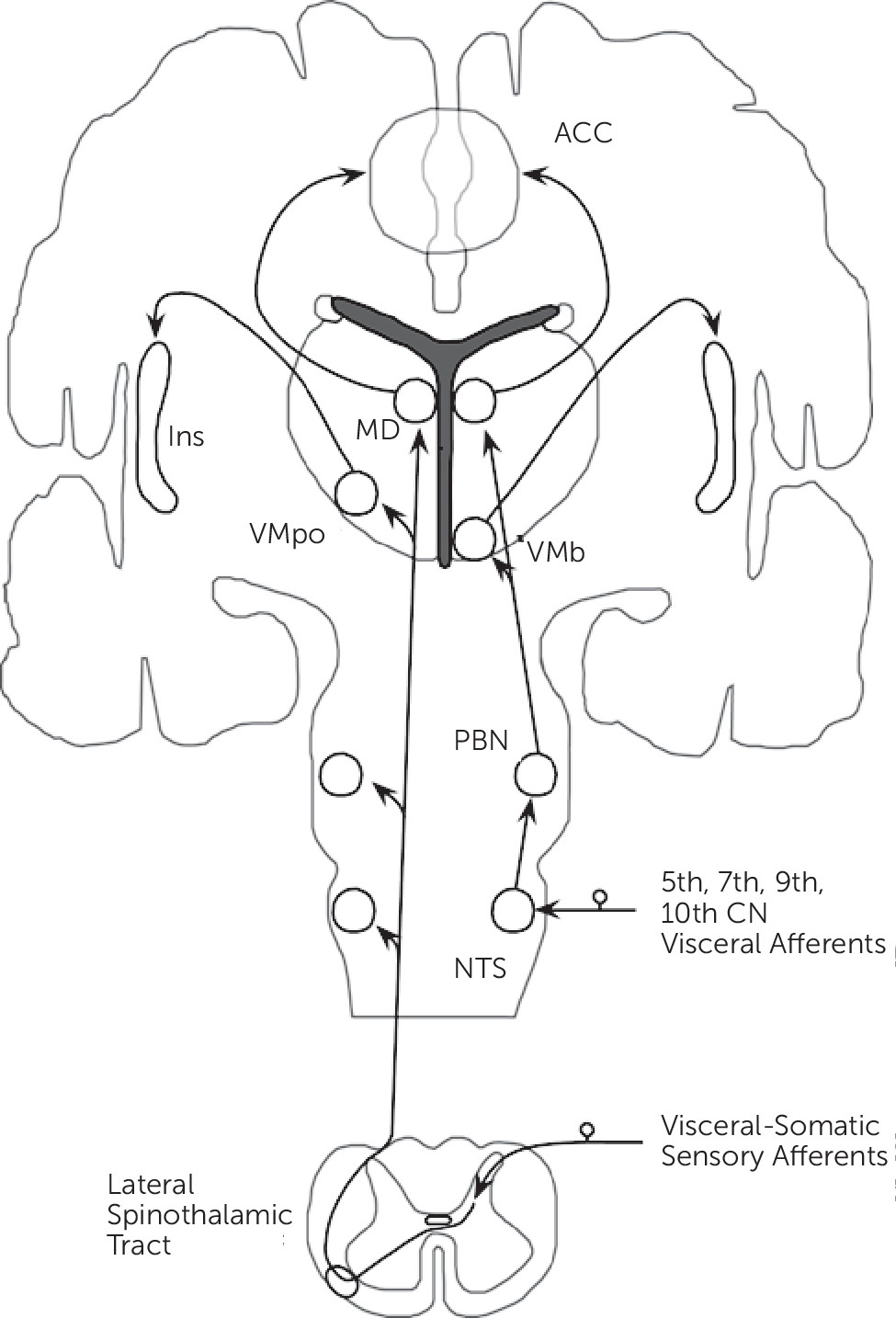
Neurobiological, noninvasive neuroimaging, and neuropathological studies have identified distributed neural networks involved in somatic and visceral sensory processing. This section focuses on sites of information convergence, and mainly details the lamina I spinothalamocortical pathway (
Figure 1).
10Small diameter sensory afferents conveying physiological information from tissues of the body (including mechanical, thermal, stress, metabolic, inflammatory, and visceral) terminate in lamina I within the dorsal horn of the spinal cord. Second-order neurons project contralaterally in the spinal cord and ascend as part of the lateral spinothalamic tract. These ascending projections, prior to terminating in the thalamus, project within the brainstem onto the nucleus tractus solitarius (NTS), parabrachial nucleus (PBN), and nucleus cuneiformis (NCF). Visceral afferents from the 5th, 7th, 9th, and 10th cranial nerves terminate on the NTS, which subsequently project onto the PBN. Lamina I spinothalamic afferents synapse topographically onto the posterior part of the ventromedial nucleus (VMpo) and the medial dorsal nucleus (MD) of the thalamus. Afferents from the PBN project onto an adjacent part of the thalamus, the basal ventral medial nucleus (VMb), and the MD nucleus. [Note: conventional spinothalamic afferents synapse ventrolaterally in the thalamus before projecting to primary somatosensory cortex (SI).] Notably, visceral-somatic afferents converge within the brainstem and the MD nucleus of the thalamus.
The VMpo and VMb thalamic nuclei project topographically onto the posterior insula, while the MD nucleus projects to the anterior cingulate cortex (ACC). Based on their afferent connections, the ACC and insula are major cortical sites of visceral-somatic processing and information convergence; the ACC and insula are reciprocally connected and these regions also connect to the orbitofrontal cortex (OFC) and the amygdala.
11 Additional components of visceral-somatic processing include SI, secondary somatosensory (SII), posterior parietal, medial and lateral prefrontal cortices, the striatum, hippocampal formation, lateral thalamus, hypothalamus, pituitary, and brainstem structures including the periaqueductal gray (PAG) and rostral ventromedial medulla (RVM). The brainstem structures form part of the descending pain neuromodulatory system, which may either inhibit or amplify visceral-somatic processing within the spinal cord.
12 Of note, while differences in the cortical-subcortical processing of visceral and somatic stimuli have been reported,
13 evidence suggests considerable cortical-subcortical and brainstem overlap.
14–17Neuroimaging Studies in Somatization Disorder, Undifferentiated Somatoform Disorder, and Somatoform Pain Disorder
Functional and structural neuroimaging studies have used fMRI, PET, SPECT, and structural MRI techniques to investigate neurocircuit abnormalities in somatization disorder, undifferentiated somatoform populations, and somatoform pain disorder.
Neuroimaging studies thus far have mainly identified striatal and amygdalar abnormalities in somatization disorder and undifferentiated somatoform disorder. Hakala and colleagues performed the first series of investigations in 10 female patients (six with somatization disorder; four with undifferentiated somatoform disorder) compared with healthy subjects using resting-state, fluorodeoxyglucose-PET, and demonstrated bilateral caudate-putamen hypometabolism in patients.
18,19 The same cohort exhibited increased bilateral caudate volumes compared with controls using a manualized MRI tracing technique.
20 In addition to the striatum, amygdalar abnormalities have also been characterized. Manualized tracings identified bilateral amygdalar volumetric reductions in 20 women with somatization disorder compared with healthy females.
21 Similarly, in an fMRI affectively valenced picture viewing task, 20 mixed somatoform disorder patients (13 undifferentiated, five somatoform pain disorder, two somatization disorder) exhibited decreased left amygdala and right parahippocampal activity during processing of emotionally valenced facial expressions compared with unrecognizable smoothed pictures.
22 Less frequently, smaller pituitary volumes were reported in somatization disorder using manualized tracings,
23 and bilateral superior temporal and left lateralized postcentral, precentral, inferior parietal, and middle occipital regional SPECT hyperperfusion was identified in patients with undifferentiated somatoform disorder compared with healthy subjects.
24Studies of patients with somatoform pain disorder commonly, though not exclusively, demonstrated increased regional central pain processing activity and reduced prefrontal cortex activity. Several investigations have compared patients with somatoform pain disorder with healthy subjects using pain provocation paradigms. In a fMRI study using a pinprick noxious stimuli task, 17 mixed-gender somatoform pain disorder patients compared with healthy subjects showed increased activity in pain processing regions including the anterior insula, hippocampus, putamen, and thalamus; in addition, somatoform pain disorder patients exhibited increased inferior parietal, temporo-occipital, lateral temporal, ventrolateral prefrontal, and dorsomedial prefrontal regional activity.
25 This somatoform pain disorder cohort compared with healthy subjects also demonstrated increased left insula and decreased bilateral temporo-occipital, superior parietal and right OFC activity during exposure to pictures and audio scenes of physical violence. Noxious thermal stimuli delivery was associated with anterior insula, parahippocampal, and amygdalar hyperactivity, along with orbitofrontal/ventromedial prefrontal hypoactivity, in 12 women with somatoform pain disorder compared with healthy subjects.
26 Decreased lateral prefrontal and increased right ACC, left thalamus and bilateral brainstem, caudate, and posterior cingulate cortex regional cerebral blood flow were also noted in a SPECT study of 10 patients with somatoform pain disorder compared with healthy subjects.
27 In parallel, an automated voxel-based structural MRI whole-brain analysis identified decreased prefrontal (ventromedial/OFC/ACC/middle frontal/superior medial), insula, parahippocampal, inferior temporal, and posterior cingulate cortex gray matter volumes.
28 Resting-state functional neuroimaging studies have reported increased brainstem, caudate, thalamus and ACC activity, and decreased lateral prefrontal activity.
29,30 In summary, neuroimaging studies suggest enhanced central pain processing activity in patients with somatoform pain disorder, whereas striatal and amygdalar dysfunction have been linked to somatization disorder and undifferentiated somatoform disorder.
A Neural Circuit Model of Somatosensory Amplification
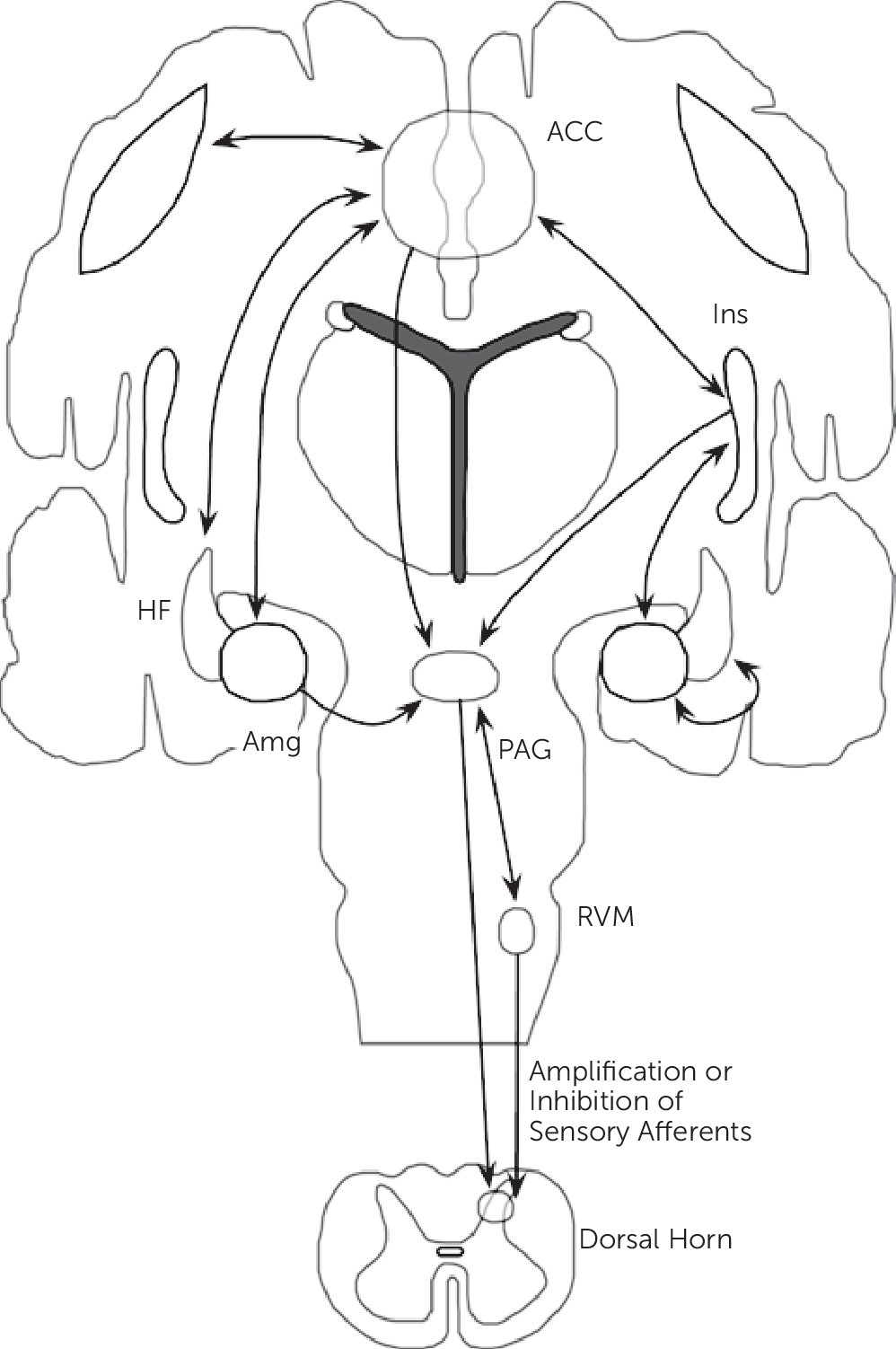
Synthesizing neuroimaging findings across somatization disorder, undifferentiated somatoform disorder, and somatoform pain disorder, and integrating these abnormalities with cognitive-affective neuroscience findings, we propose that aberrant circuit interactions across large-scale neural systems mediating visceral-somatic perception, emotional processing/awareness, and cognitive control serve critical roles in the neurobiology of somatosensory amplification. Important brain regions linked to somatosensory amplification include the ACC, insula, amygdala, hippocampal formation, and striatum among other regions (
Figure 2).
The lamina I spinothalamic pathway allows for the integration of visceral-somatic information within the brainstem, thalamus, and cortex. Visceral-somatic information converges cortically within the ACC and insula. The ACC has been classically subdivided into a subgenual/pregenual affective component and a dorsal ACC/anterior middle cingulate cortex (aMCC) cognitive component.
70 Nociceptive studies across visceral-somatic stimuli elicit robust cingulate gyrus activations; visceral stimuli frequently activate pregenual and dorsal ACC, whereas noxious cutaneous stimuli more robustly activate the MCC. Uniquely positioned for integration is the dorsal ACC/aMCC where affectively ladened information, visceral-somatic processing, motivated behavior, and cognitive control converge. Shackman and colleagues
71 recently proposed this region as a critical integrator of negative affect, pain, and cognitive control. ACC-subcortical connections include the nucleus accumbens/ventral caudate, ventral globus pallidus, and MD, and ventral anterior thalamic nuclei; cortico-cortical connections occur between the ACC and the dlPFC, insula, OFC, amygdala, and hippocampal formation.
11 ACC/aMCC functional and structural abnormalities have been reported in somatoform pain disorder,
27,28 and this region is involved in negative expectation bias,
36,38,39 negative attentional bias,
40,43,44 alexithymia,
52–55,57,58,60,61 and negative affect modulation of visceral-somatic processing.
64,65,68 Cortical ACC‒dlPFC interactions may mediate negatively valenced, visceral-somatic forecasting associated with catastrophizing,
47 and negative attentional bias, whereas ACC‒OFC connections may mediate top-down influences of negative expectation and negative mood hierarchically. Caudate-putamen components of the ACC, dlPFC, and OFC cortical-subcortical circuits potentially relate more specifically to somatization and undifferentiated somatoform disorders.
18–20Apart from the ACC, the insula is also likely a critical region for somatosensory amplification. The posterior insula receives somatosensory, nociceptive/thermoceptive, and visceral information from the thalamus. Classic intraoperative electrical stimulation studies of the posterior insula performed by Penfield verified that many visceral sensations including gurgling, burning, rising/rolling sensations, and nausea can be produced through posterior insula activations.
72 A.D. Craig has specifically suggested that the posterior insula provides an interoceptive representation of the physiological condition of the body.
10 The mid-insula is considered an integrative zone where affectively and motivationally valenced information from the ACC, amygdala, and OFC influence sensory processing. The integration of visceral-somatic, affective, and motivational information converges onto the anterior insula, and together with the ACC, the anterior insula (right>left) has been linked to emotional awareness.
73 Interestingly, the insula and ACC share large spindle-shaped neurons, termed von Economo neurons, linked to social-emotional cognition. Differential insular activity occurs in somatization disorder,
74 undifferentiated somatoform disorder,
74 and somatoform pain disorder,
25,26,28,74 and the insula is involved in negative expectation bias,
37–39 alexithymia,
54,62 and the modulation of visceral-somatic processing by negative emotion.
64,65,69 Opioid system activity may potentially drive insula and ACC related negative expectation responses within somatoform illness,
39 particularly in the context of negative emotion.
Somatosensory amplification may involve a bidirectional pattern of insula and amygdala activity. Patients with somatization disorder, undifferentiated somatoform disorder, and somatoform pain disorder
22 exhibit insula and amygdala hypoactivity to external (environmental) emotionally valenced stimuli. Amygdalar and insular hypoactivity was also observed in subjects with alexithymia exposed to extrinsic emotionally valenced stimuli.
61,62 Conversely, delivery of self-oriented, bodily-related stimuli (i.e., tactile) increased amygdala and insula activity in patients with somatoform pain disorder,
25–27,30 and these regions also showed hyperactivity in studies of negative attentional bias.
40–44 These findings suggest that somatosensory amplification may be partially the result of selective heightened attention and salience for bodily sensations (internal states) and parallel under-processing of external emotionally valenced information. Tryptophan depletion studies suggest serotonergic dysfunction may potentially mediate this bidirectional aberrant activity pattern.
The hippocampal formation (including the parahippocampal gyrus) also demonstrated abnormal activity in somatization disorder,
22 undifferentiated somatoform disorder,
22 and somatoform pain disorder patients.
22,25,26 Similarly, studies of negative expectation bias
31–35 reported increased hippocampal formation activity. Using the Gray-McNaughton theory,
75 which posits a role for the hippocampal formation in responding to aversive information in the context of behavioral conflict (including uncertainty, novelty, and contextual processing), these studies suggest an amplifying role for the hippocampal formation in visceral-somatic processing during uncertainty; this effect may be mediated by enhanced insula and ACC activity.
31The ACC, insula, amygdala, and hippocampal formation are all part of the descending pain modulatory system, which also includes multiple brainstem structures (PAG, NCF, and RVM).
12 Reciprocal connections between the ACC and the amygdala, OFC, dlPFC, insula, and hippocampal formation position the ACC as a key cortical modulatory region of PAG activity (both directly and indirectly though ACC-amygdala-PAG connections). Efferent connections between the ACC and the PAG have been described in mammals, though DTI failed to reliably identify human ACC-PAG connections
76 suggesting further anatomical clarification is necessary. Nonetheless, the PAG is connected to and modulated by the amygdala, insula and multiple prefrontal cortical regions as well.
76 Within the brainstem, the PAG is interconnected with the RVM and NCF. Descending RVM and PAG projections can either amplify or inhibit afferent sensory processing within the dorsal horn of the spinal cord. These cortico-brainstem connections likely play a role in somatosensory amplification and future research will help determine if intrinsic brainstem abnormalities (independent of cortical, top-down influence) are also involved in the pathophysiology of somatosensory amplification.
Having delineated a neurocircuit framework for somatosensory amplification, it is important to emphasize that in a given subject or patient group, dysfunction in distinct nodes or levels of these hierarchical cortical-subcortical-brainstem-spinal cord circuits may occur. One patient may have amplification of visceral-somatic processing driven by enhanced entry-level afferent information within the dorsal horn of the spinal cord or brainstem nuclei, whereas another individual may have amplification driven by cortical-subcortical mediated cognitive and affective processes.
Additional Considerations–Inflammation, Stress-Mediated Neuroplastic Change and Autonomic Imbalance
Although this article focuses on neurocircuit disturbances, it is important to introduce roles for inflammation, aberrant neuroplastic change, and autonomic imbalance in the pathophysiology of somatosensory amplification. Recent research suggests that proinflammatory states modulate frontolimbic circuits to alter affective and cognitive elements of visceral-somatic processing. Chronic interpersonal stress, prevalent in patients with somatoform disorders, has been associated with enhanced inflammatory leukocyte response to microbial challenge,
77 and women with elevated levels of circulating cytokines report poorer health despite controlling for physical health and diagnosis.
78 Furthermore, pain catastrophizing following noxious stimuli delivery has been associated with interleukin-6 (IL-6) reactivity.
79 A series of studies by Harrison and colleagues explored the modifying effects of systemic inflammation on neural activity.
80,81 In a double-blind, randomized study using typhoid injection compared with placebo, individuals who received a typhoid injection reported worsening mood and demonstrated increased systemic IL-6 levels. In the typhoid injected group, enhanced subgenual ACC (sgACC) activity was associated with dysphoric mood; decreased functional connectivity of the sgACC to the amygdala, medial prefrontal cortex, and nucleus accumbens was modulated by peripheral IL-6 levels.
80 In this same cohort, self-reported fatigue following injection correlated with bilateral mid/posterior insula and left ACC activity.
81 These studies suggest a neuromodulatory role for circulating cytokines in the emergence of dysphoric mood and fatigue.
Aberrant neuroplastic change and impaired development following early-life interpersonal stress, including sexual/physical abuse and neglect, are additional elements potentially mediating the emergence of somatic symptoms. Patients with somatoform illness frequently experience early-life trauma. Animal models of chronic stress suggest that the medial prefrontal cortex (including the ACC), hippocampus, and amygdala undergo neuroplastic changes in response to prolonged stress.
82 The hippocampal CA3 region and medial prefrontal cortex exhibit dendritic spine density reductions following repeated stress; amygdalar experience-dependent changes, although less well studied, have also been described. Recent large-scale volumetric analyses in human subjects observed ACC, OFC, insula, hippocampal, and caudate gray matter reductions associated with a history of childhood trauma.
83 Early-life interpersonal stress also predisposes to insecure adult attachment. Individuals with insecure attachment display amygdala and striatal dysfunction,
84 and abnormal hypothalamic-pituitary-adrenal axis function.
85 Strikingly, these findings overlap with brain regions theorized to play a role in abnormal somatosensory amplification.
An imbalance of the autonomic nervous system has also been reported in somatoform disorders.
86–90 Rief and colleagues characterized elevated morning heart rates and cortisol levels, along with persistent heart rate increases (failed habituation) during an emotionally valenced word viewing task in patients with prominent somatization compared with healthy controls
90; a similar cohort of patients exhibited persistently elevated heart rates during rest following completion of an attentional task.
89 Patients with somatization disorder displayed increased heart rate and decreased baroreceptor sensitivity during autonomic testing.
86 Patients with somatoform symptoms also exhibited reduced heart rate variability during affectively valenced facial viewing
88 and pain processing paradigms.
87 These findings suggest a static imbalance of increased sympathetic and decreased parasympathetic tone. Thayer and colleagues proposed an important role for the central autonomic network (CAN), which includes frontolimbic, insular and brainstem structures, in the pathobiology of autonomic imbalance facilitating noxious psychosomatic experiences.
91 Importantly, the CAN converges with the theorized neurocircuitry of somatosensory amplification.
Therapeutic Implications
Having proposed prominent prefrontal dysfunction (i.e., ACC, dlPFC, OFC) in the pathophysiology of somatosensory amplification, several therapeutic implications emerge. Noninvasive and invasive neuromodulation, particularly rapid transcranial magnetic stimulation (rTMS) and deep brain stimulation (DBS), have been investigated in the treatment of neuropsychiatric disorders. Modulation of dlPFC activity, in conjunction with trans-synaptic sgACC modulation, has been suggested as the mechanism for therapeutic efficacy of rTMS in the treatment of major depressive disorder.
92 Future investigations in somatoform illness should evaluate dlPFC neuromodulation to potentially improve pain catastrophizing, negative attentional bias, and negative affective disturbances. DBS targeting sensory thalamus and periventricular/periaqueductal gray matter have been used to treat chronic intractable pain syndromes for the past half-century. Interestingly, ventral caudal thalamic stimulation for chronic pain treatment modulated perigenual and dorsal ACC activity,
93 and ventral PAG stimulation enhanced parasympathetic activity.
94 For patients with disabling, chronic somatoform pain disorders, consideration should be given to investigating the PAG, dorsomedial thalamus, and dorsal ACC as potential therapeutic targets.
Prominent roles for the monoamine and opioid neurotransmitter systems in negative attentional bias, negative expectation, and affective disturbances in somatosensory amplification are consistent with the pharmacologic evidence in somatoform disorders. A meta-analysis of antidepressants (mainly tricyclic and selective serotonin reuptake inhibitors) compared with placebo for the treatment of somatoform pain disorder showed significant decreased pain intensity following antidepressant use.
95 A more recently conducted randomized, double-blind placebo-controlled trial of fluoxetine in patients with somatoform pain disorder compared with controls demonstrated drug-related analgesia, with the greatest therapeutic efficacy in patients with comorbid depression.
96 Negative expectation, particularly the nocebo effect, has been linked to decreased opioid and dopaminergic neurotransmitter activity,
39 and this finding may be particularly noteworthy in the context of an association between severe somatization and opiate drug misuse.
Empirical evidence supports using cognitive-behavioral therapy (CBT) for the treatment of somatoform disorders. Randomized controlled trials investigating the effectiveness of CBT for somatization disorder, undifferentiated somatoform disorder, and somatoform pain disorder have demonstrated improvements in physical symptom severity and level of functioning.
97,98 The particular mechanisms utilized in CBT [which include modifying physiological arousal, attention, attributional processes, and cognitive distortions (i.e., catastrophizing)] target many of the theorized cognitive and affective modifiers of visceral-somatic processing in somatosensory amplification.
Emerging data for mindfulness-based techniques (MBT) also suggest some promise in targeting the circuitry of abnormal somatosensory amplification. MBT involve training in specific meditative practices that encourage moment-to-moment, nonjudgmental, nonreactive awareness. Through such training, it has been proposed that a distributed, large-scale network (including the dorsal ACC, dlPFC, and anterior insula among other regions) are functionally recruited to guide therapeutic changes in neural systems underlying catastrophizing and affect-biased attention.
99 Interestingly, the most widely cited brain areas of activity and morphological change during and in response to MBT have been the ACC, dlPFC, anterior insula, and hippocampus.
99 MBT have been reported to reduce disability pensions in patients with somatization disorder
100 and should be further investigated in patients with somatoform illness.
Limitations and Future Directions
There are several important limitations to address regarding this theoretical neural circuit framework for somatosensory amplification. Even though this approach integrates somatoform disorder neuroimaging abnormalities with related findings in cognitive-affective neuroscience, investigations probing cognitive and affective modifiers of visceral-somatic processing in somatoform disorder populations are necessary to validate the proposed model. In addition, there are limited, underpowered somatoform disorder-specific neuroimaging studies to date, and case-control studies with increased sample sizes are needed to ensure the reliability of the framework, and clarify important concepts such as the role of gender in the pathophysiology of somatosensory amplification. Several somatoform disorder visceral-somatic symptoms (i.e., noncardiac chest pain, breathlessness, dizziness) are also understudied using brain imaging techniques and, thus, not fully integrated into the current framework; these symptoms, however, likely map onto the neurocircuitry in topographic fashion with similar influences from cognitive and affective amplifiers. Future research is necessary to also clarify intra and intercircuit functional connectivity patterns and to delineate common and disorder-specific circuit abnormalities across somatoform disorder subtypes. In addition, this framework focuses on central modifiers of visceral-somatic processing, however, primary end organ dysfunction (i.e., abnormal serotonergic transmission in the gut wall) should be explored for potential additive or synergistic roles in the pathophysiology of symptom amplification. Future research will also incorporate genetic-epigenetic influences, and the neuroimaging based systems-level approach taken in this article should be further refined with other systems-level research modalities including electrophysiology techniques (i.e., event-related potentials). Systems-level measures of brain function, which may be associated with complex mental states, are also not necessarily inherently causative of specific abnormal symptoms; this highlights the need for integrative multilevel research investigations in somatosensory amplification. Lastly, while critical neurocircuit regions were identified, it will be necessary to further investigate ACC (subgenual, perigenual, dorsal) and insula (anterior-midposterior) subregion involvement and directionally of abnormal activation patterns in the pathophysiology of somatosensory amplification.