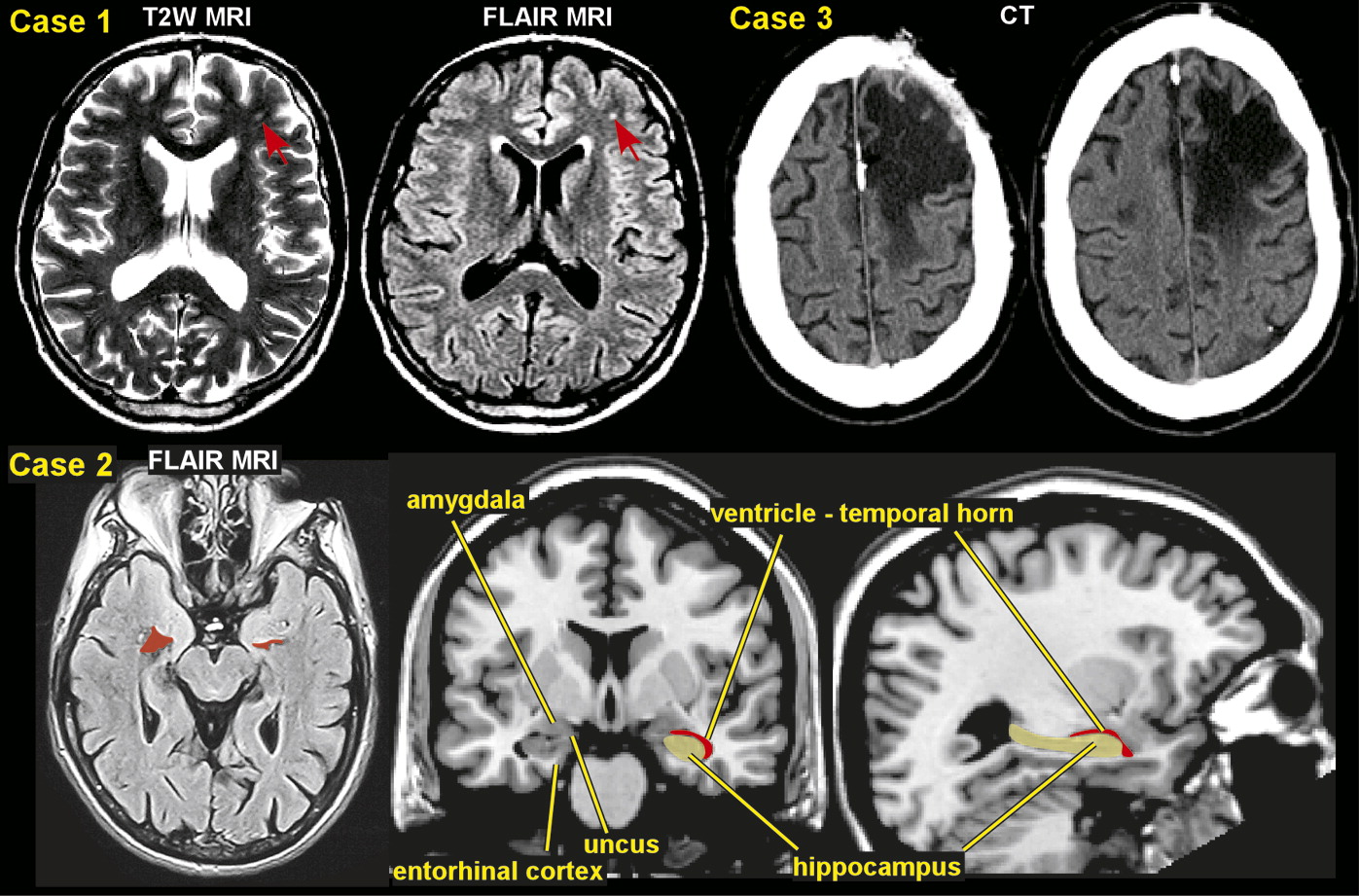
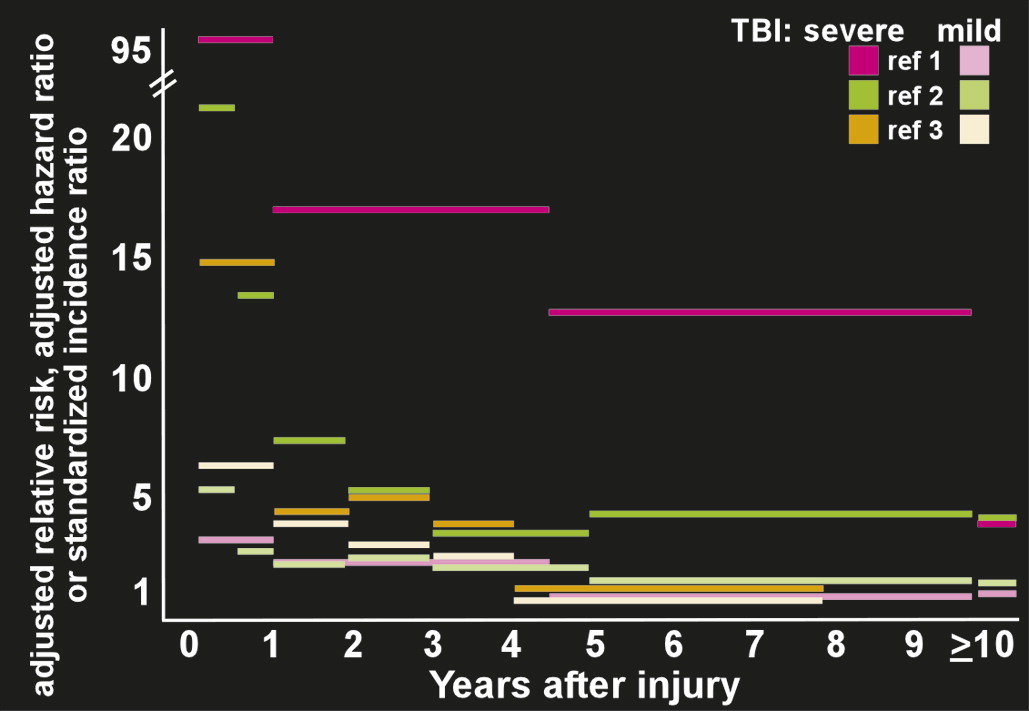
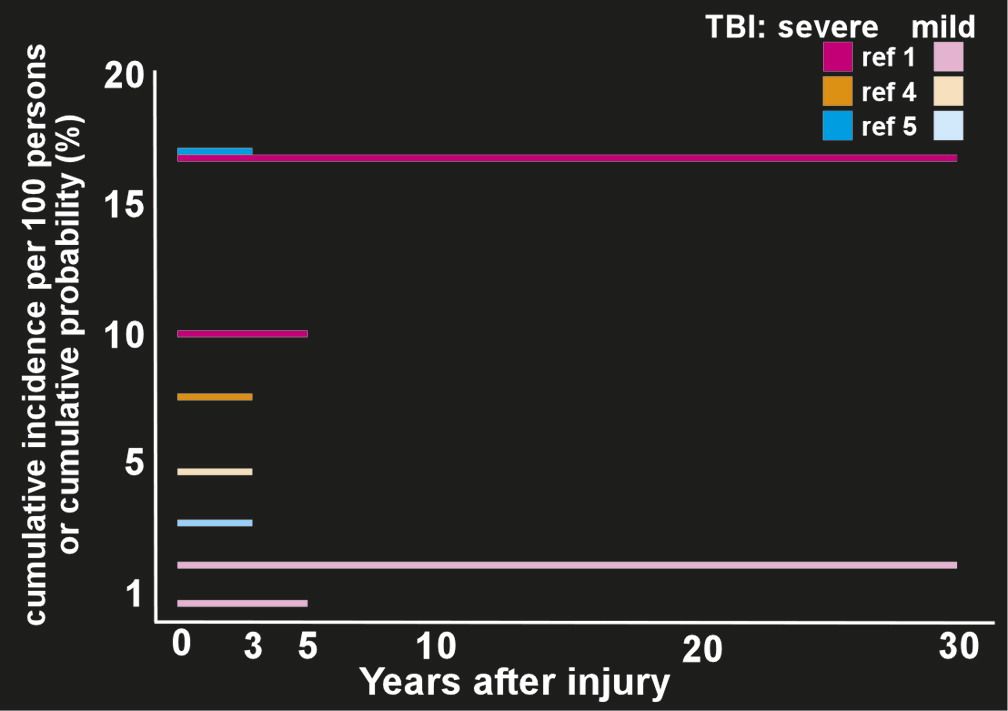
The development of PTE after a latent period is an example of human epileptogensis, whereby a nonepileptic brain undergoes molecular and cellular alterations after a brain insult, which increases its excitability.
7–9 This eventually leads to the occurrence of recurrent spontaneous seizures. Penetrating brain injury produces a cicatrix (scar) in the cortex and is associated with an increased risk of PTE of approximately 50%.
29 The initial disruption of tissue may be compounded by ischemia and hemorrhage. Nonpenetrating injuries, including focal contusions and intracranial hemorrhages, are associated with a risk of PTE of up to 30%. In this setting, the mechanism of epileptogenesis may be partly related to the toxic effects of hemoglobin breakdown products on neuronal function.
8,36 Closed head injuries produce diffuse axonal injury with shearing of axons, diffuse edema, and ischemia. The most common locations are the gray matter-white matter junction particularly in the frontal and temporal areas.
37 This process leads to the release of excitatory amino acids, cytokines, bioactive lipids, and other toxic mediators causing secondary cellular injury.
7–9 Epileptogensis is thought to be initiated by specific types of cell loss and neuronal reorganization, which results not only in enhanced excitation, but also in decreased inhibition, predisposing to hypersynchronization.
38 Brain injuries also lead to upregulation of proinflammatory cytokines and activated immune responses to further increase seizure susceptibility, promote neuronal excitability, and impair blood–brain barrier (BBB) integrity.
39,40 Early changes include the induction of immediate early genes and post-translational modifications of neurotransmitter receptor and ion channel/transporter proteins.
41 Within days, neuronal death, initiation of an inflammatory cascade, and new gene transcription has been reported to occur.
42 Later (days to weeks) anatomic changes include axonal sprouting and dendritic modifications. For example, mossy fiber sprouting can be observed in chronic epileptic brain.
43 Over time, seizure threshold is lowered by a growing increase in excitability, and the risk of a seizure increases.
44The process by which trauma to the brain tissue leads to hyperexcitability, hypersynchronization, and development of recurrent seizures is relatively unknown, but there are several theories as to potential mechanisms based on experimental data, primarily from animal models of PTE. Animal models of TBI (mostly performed in rodents) are divided into focal brain injury, diffuse brain injury, mixed models of focal and diffuse brain injury, and models of repetitive concussive injury.
45,46 These models utilize a direct impact on the epidural space on the brain tissue.
45 A large number of studies have used lateral fluid-percussion or central fluid-percussion TBI models that produce both gray matter and white matter injury.
47 Late spontaneous seizures (PTE) have consistently been reported in the rat fluid-percussion TBI model, which is the most commonly used model of human closed head TBI.
47 The physiological effects from TBI are considered to consist of several phases including primary injury, evolution of primary injury, secondary injury, and regeneration.
45,47 Multiple mechanisms are likely activated simultaneously or sequentially by brain trauma. Animal studies demonstrate that a large number of molecular and cellular alterations occur during the latent period that could be related to epileptogenesis, including cell death, gliosis, neurogenesis, axonal and dendritic plasticity, rearrangement of the extracellular matrix, and angiogenesis.
47