Psychobiological Mechanisms of Resilience and Vulnerability: Implications for Successful Adaptation to Extreme Stress
Abstract
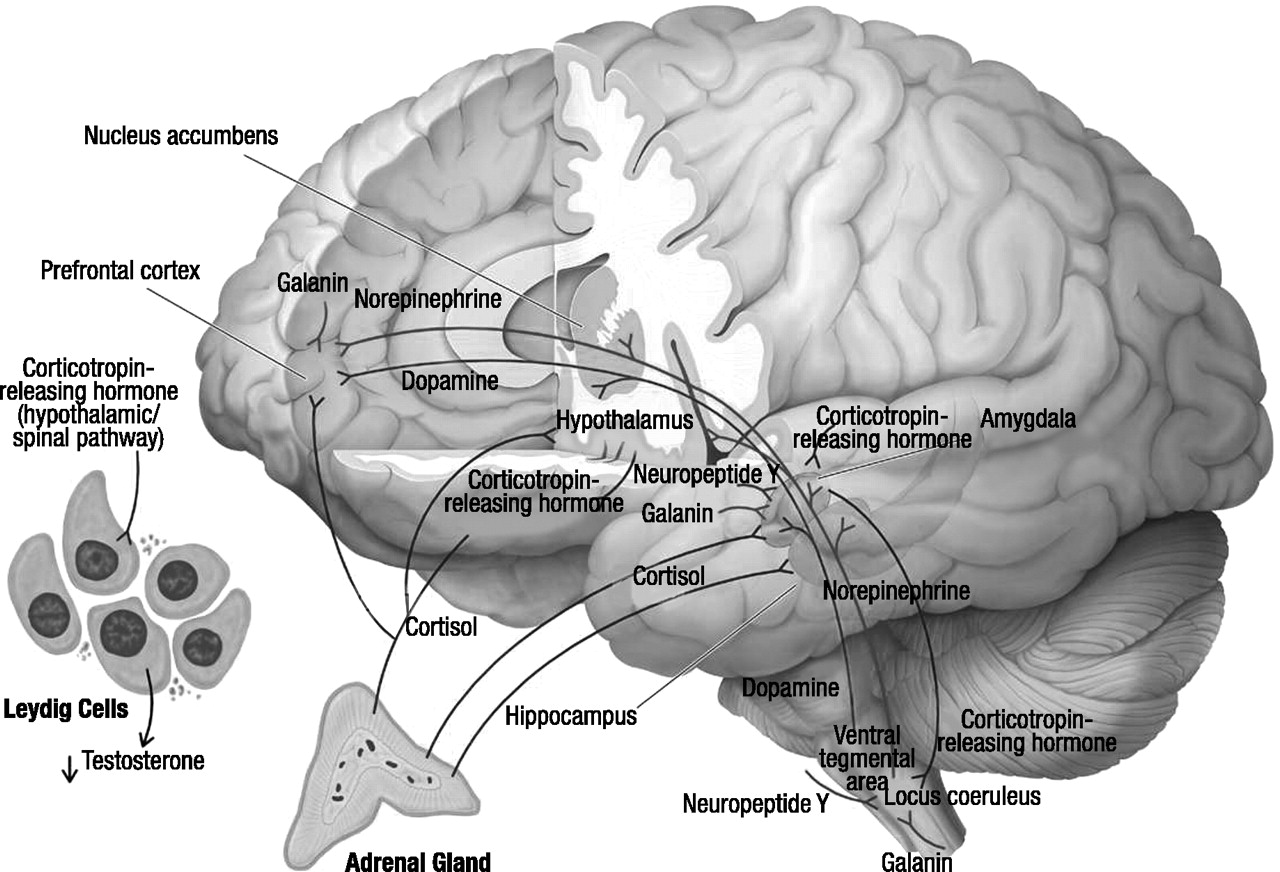
Cortisol and dehydroepiandrosterone
CRH
Locus coeruleus–norepinephrine system
Neuropeptide Y
Galanin
Dopamine
Serotonin
Benzodiazepine receptors
Gonadal steroids
Testosterone
Estrogen
Resilience and vulnerability to stress
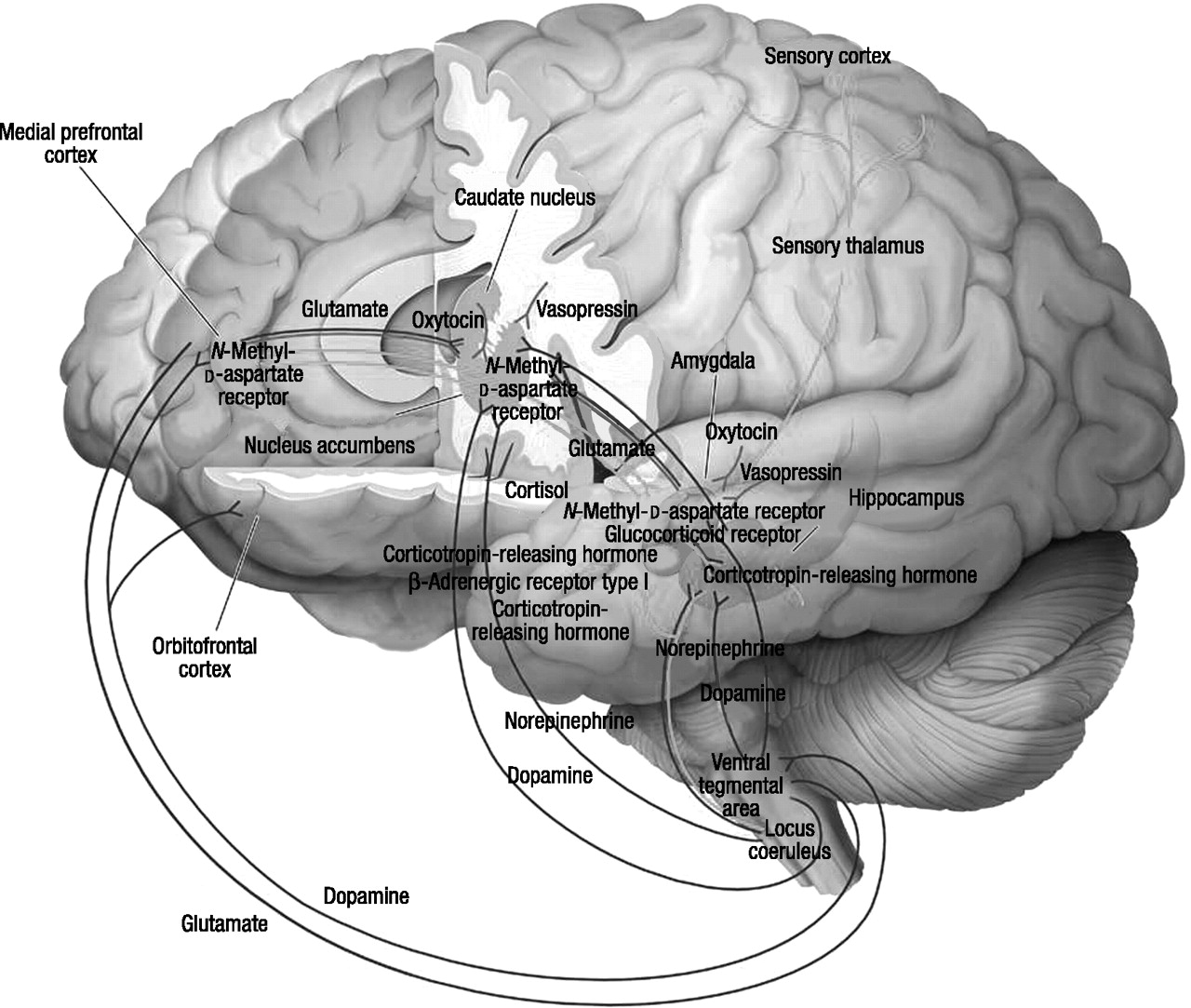
Reward, fear conditioning, and social behavior
Regulation of reward
The neural mechanisms of anxiety and fear
Fear conditioning
Reconsolidation
Extinction
The neural basis of social behavior
Future research directions
| Neurochemical | Acute Effects | Brain Regions | Key Functional Interactions | Association With Resilience | Association With Psychopathology |
|---|---|---|---|---|---|
| Cortisol | Mobilized energy, increased arousal, focused attention, fear memory formation, fear learning | Prefrontal cortex, hippocampus, amygdala, hypothalamus | Increases amygdala corticotropin-releasing hormone (CRH), increases hypothalamic CRH | Stress-induced increase constrained by negative feedback by means of glucocorticoid receptor and mineral corticoid receptors | Unconstrained release leads to hypercortisolemia-depression, hypertension, osteoporosis, insulin resistance, coronary vascular disease; overconstrained release leads to hypocortisolemia, seen in some PTSD patients |
| Dehydroepian-drosterone (DHEA) | Counteracts deleterious effects of high cortisol neuroprotection; has positive mood effects | Largely unknown; hypothalamus | Antiglucocorticoid actions | High DHEA-cortisolratios may have preventive effects regarding PTSD and depression | Low DHEA response to stress may predispose to PTSD and depression and the effects of hypercortisolemia |
| CRH | Activated fear behaviors, increased arousal, increased motor activity, inhibited neurovegetativefunction, reduced rewardexpectations | Prefrontal cortex, cingulate cortex, amygdala, nucleus accumbens, hippocampus, hypothalamus, bed nucleus of the stria terminalis, periaqueductal gray matter, locus coeruleus, dorsal raphe | CRH-1 receptoranxiogenic, CRH-2 receptor anxiolytic, increases cortisol and DHEA, activateslocus coeruleus-norepinephrine system | Reduced CRH release, adaptive changes in CRH-1 and CRH-2 receptors | Persistently increased CRH concentration may predispose to PTSD and major depression; may relate to chronic symptoms of anxiety, fear, and anhedonia |
| Locus coeruleus-norepinephrine system | General alarm function activated by extrinsic and intrinsic threat; increased arousal, increased attention, fear memory formation, facilitated motor response | Prefrontal cortex, amygdala, hippocampus, hypothalamus | Activates sympathetic axis, inhibits parasympathetic outflow, stimulates hypothalamic CRH | Reduced responsiveness of locus coeruleus-norepinephrine system | Unrestrained functioning of locus coeruleus-norepinephrine system leads to chronic anxiety, hypervigilance, and intrusive memories; some patients with PTSD, panic disorder, and major depression show evidence of heightened locus coeruleus-norepinephrine activity |
| Neuropeptide Y | Anxiolytic; counteracts thestress-related effects of CRH and the locus coeruleus-norepinephrine system; impairs fear memory | Amygdala, hippocampus, hypothalamus, septum, periaqueductal gray matter, locus coeruleus | Reduces CRH-related actions at amygdala, reduces rate of firing of locus coeruleus | Adaptive increase in amygdala neuropeptide Y is associated with reduced stress-induced anxiety and depression | Low neuropeptide Y response to stress is associated with increased vulnerability to PTSD and depression |
| Galanin | Anxiolytic; counteracts the stress-induced effects of the locus coeruleus-norepinephrine system; impairs fear conditioning | Prefrontal cortex, amygdala, hippocampus, hypothalamus, locus coeruleus | Reduces the anxiogenic effects of locus coeruleus-norepinephrine system activation | Adaptive increase in amygdala galanin is associated with reduced stress-induced anxiety and depression | Hypothesized low galanin response to stress is associated with increased vulnerability to PTSD and depression |
| Dopamine | High prefrontal cortex and low nucleus accumbens dopamine levels are associated withanhedonic and helpless behaviors | Prefrontal cortex, nucleus accumbens, amygdala | Reciprocal interactions between cortical and sub-cortical dopamine systems | Cortical and subcortical dopamine systems remain in optimal window of activity to preserve functions involvingreward and extinction of fear | Persistently high levels of prefrontal cortical and low levels of subcortical dopamine activity are associated with cognitive dysfunction and depression; persistently low levels of prefrontal cortical dopamine are associated with chronic anxiety and fear |
| Serotonin (5-HT) | Mixed effects: 5-HT stimulation of 5-HT2 receptorsis anxiogenic; 5-HT stimulation of 5-HT1A receptors is anxiolytic | Prefrontal cortex, amygdala, hippocampus, dorsal raphe | High levels of cortisol decrease in 5-HT1A receptors | High activity of postsynaptic 5-HT1A receptors may facilitate recovery | Low activity of postsynaptic 5-HT1A receptors may predispose to anxiety and depression |
| Benzodiazepine receptors | Acute stress down-regulation of cortical benzodiazepinereceptors | Prefrontal cortex, hippocampus | May be relationship between decreased 5-HT1A and decreased benzodiazepinereceptor function | Resistance to stress-induced down-regulation of benzodiazepine receptors | Decreased cortical benzodiazepine receptors are associated with panic disorder and PTSD |
| Testosterone | Stress-induced decrease in assertive behavior and increase in depression | Hypothalamus | CRH decreases testosterone levels | Increase in testosterone may promote increased energy and active coping and reduce depression symptoms | Decreased CSF testosterone levels are found in PTSD; testosterone supplementation is helpful for depressed men with low testosterone levels |
| Estrogen | Acute increasesin estrogen may dampen hypothalamic-pituitary-adrenal (HPA) and norepinephrine responses to stress | Hypothalamus, hippocampus | Estrogen increases function of benzodiazepine receptors and decreases function of 5-HT1A receptors | Short-term increases in estrogen may attenuate effects of stress-induced HPA axis and noradrenergic system activation | Long-term increases in estrogen may down-regulate 5-HT1A receptors and increase risk or depression and anxiety |
| Mechanism | Neurochemical Systems | Brain Regions | Association With Resilience | Association With Psychopathology |
|---|---|---|---|---|
| Reward | Dopamine, dopamine receptors, glutamate, N-methyl-d-aspartic acid (NMDA) receptors, γ-aminobutyric acid (GABA), opioids, cAMP response element binding protein, ΔFosB | Medial prefrontal cortex, nucleus accumbens, amygdala, hippocampus, ventral tegmental area, hypothalamus | Acute and chronic stress do not produce impairment in neurochemical or transcription factor-mediated reward | Stress-induced reduction in dopamine and increases in cAMP response-element binding protein transcription produces a dysfunction in reward circuitry leading to anhedonia and hopelessness |
| Pavlovian (cue-specific) fear conditioning | Glutamate, NMDA receptors; voltage-gated calcium channels | Medial prefrontal cortex, sensory cortex, anterior cingulate, dorsal thalamus, lateral amygdala, central nucleus of amygdala | Adaptive association between conditioned stimuli and unconditioned stimuli does occur; fear responses are circumscribed; this may be due to functional differences in NMDA receptors and voltage-gated calcium channels; treatment with an NMDA receptor agonist (memantine) or voltage-gated calcium channel antagonists (verapamil and nimodipine) may attenuate acquisition of fear | May account for common clinical observation in panic disorder, PTSD, and depression that overgeneralization of sensory and cognitive stimuli associated with or resembling the original trauma elicits panic attacks, flashbacks, and autonomic symptoms |
| Inhibitory avoidance (contextual fear) | Norepinephrine/β-adrenergic receptor, cortisol/glucocorticoid receptor, corticotropin-releasing hormone (CRH), GABA, opioid, acetylcholine | Medial prefrontal cortex, basal lateral amygdala hippocampus, bed nucleus of the stria terminalis, entorhinal cortex | Reduced stress-induced release of CRH, cortisol, and norepinephrine decreases fear memory consolidation; CRH antagonists and β-adrenergic receptor antagonists may have preventive effects | Excessive stress-mediated release of CRH, cortisol, and norepinephrine will facilitate development of indelible fear memories; chronic anxiety and depressive symptoms may result from excessive contextual fear conditioning |
| Reconsolidation | Glutamate, NMDA receptors, norepinephrine, β-adrenergic receptors, cAMP response-element binding protein | Amygdala, hippocampus | The lability of the memory trace allows a reorganization of original memory that is less traumatic and symptom producing; treatment with NMDA receptor and β-adrenergic receptor antagonists after memory reactivation may reduce the strength of the original traumatic memory | Repeated reactivation and reconsolidation may further strengthen the memory trace and lead to persistence of trauma-related symptoms |
| Extinction | Glutamate, NMDA receptors, voltage-gated calcium channels, norepinephrine, dopamine, GABA | Medial prefrontal sensory cortex, amygdala | An ability to quickly attenuate learned fear through a powerful extinction process and an ability to function more effectively in dangerous situations may be due to inhibition of amygdala activity mediated by the medial prefrontal cortex | Failure in neural mechanisms of extinction may relate to persistent traumatic memories, reexperiencing symptoms, autonomic hyperarousal, and phobic behaviors |
Footnote
References
Information & Authors
Information
Published In


History
Authors
Metrics & Citations
Metrics
Citations
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
For more information or tips please see 'Downloading to a citation manager' in the Help menu.
View Options
View options
PDF/EPUB
View PDF/EPUBGet Access
Login options
Already a subscriber? Access your subscription through your login credentials or your institution for full access to this article.
Personal login Institutional Login Open Athens loginNot a subscriber?
PsychiatryOnline subscription options offer access to the DSM-5-TR® library, books, journals, CME, and patient resources. This all-in-one virtual library provides psychiatrists and mental health professionals with key resources for diagnosis, treatment, research, and professional development.
Need more help? PsychiatryOnline Customer Service may be reached by emailing [email protected] or by calling 800-368-5777 (in the U.S.) or 703-907-7322 (outside the U.S.).