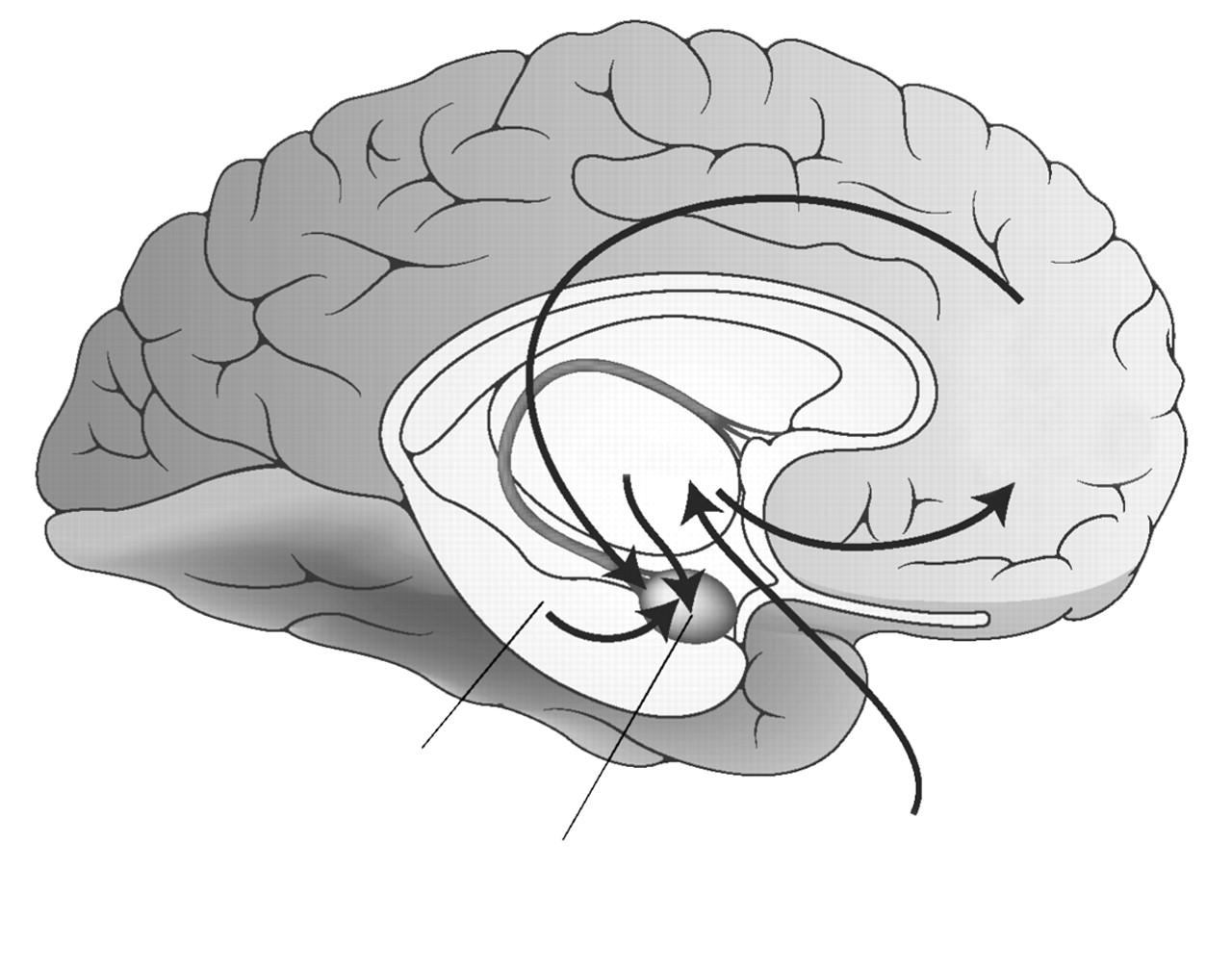
Neurocircuitry models of PTSD (
12) focus on the central role of the amygdala in directing an individual’s response to perceived danger through its reciprocal connections with the hippocampus, medial prefrontal cortex, and other cortical areas associated with higher cognitive functions. This model emphasizes a lack of adequate top-down governance over the amygdala by medial prefrontal cortex (specifically, the rostral portion of anterior cingulate cortex [13] and the hippocampus, resulting in hyperresponsivity within the amygdala to threat-related stimuli. The lack of habituation to repetitive stimuli that do not signal imminent threat and that characterizes PTSD is attributed to inadequate influence of the anterior cingulate cortex on the amygdala. Another hallmark of PTSD, the overgeneralization of fear responding to nonthreatening stimuli, is associated with decreased hippocampal function.
Structural imaging findings
Morphometric MRI studies of PTSD have focused primarily on the hippocampus, in light of its known susceptibility to stress-induced damage via the action of glucocorticoids and excitatory amino acids (
14,
15). Two studies have reported reduced hippocampal volumes in Vietnam combat veterans with PTSD (
16,
17). In one study, total hippocampal volume was inversely correlated with the extent of combat exposure and PTSD symptom severity (
17).
Three recent studies investigating potential brain alterations in children and adolescents with PTSD failed to find differences in hippocampal volumes between patients with PTSD and healthy control individuals (
18–
20). However, two of these studies reported that children with PTSD had smaller total brain and cerebral volumes (
19,
20), suggesting a more generalized effect of traumatic stress in early development. De Bellis et al. (
21) also demonstrated larger superior temporal gyrus volumes and a loss of the normal asymmetry pattern in a pediatric sample with PTSD. Abnormalities of the superior temporal gyrus, implicated in verbal and nonverbal auditory processing, may reflect abnormal developmental changes as the result of chronic childhood maltreatment resulting in PTSD.
Contrary to the pediatric findings, in mMRI studies of adults with PTSD resulting from childhood abuse, investigators have reported similar hippocampal volumetric differences to those studies examining samples with PTSD resulting from traumatic exposure in adulthood. Several studies now confirm that adult survivors of childhood sexual or physical abuse have smaller hippocampal volumes than healthy comparison samples (
22–
24,
25•). In summary, the results of studies of adults with PTSD with childhood or adult traumatic exposure support an association between reduced hippocampal volume and PTSD. However, the negative findings reported in pediatric studies suggest that hippocampal atrophy may develop in a progressive fashion over time. In a prospective study designed to test this hypothesis in an adult population, Bonne et al. (
26) found that individuals who developed PTSD as a result of a traumatic exposure did not differ from those who did not develop PTSD in hippocampal volume at baseline, or at 6 months. The results of this study suggest that significant brain changes, particularly in the hippocampus, do not occur within 6 months of developing PTSD symptoms in response to an acute traumatic event. A recent study of adult male monozygotic twins discordant for combat exposure indicated that smaller hippocampal volumes may represent a risk factor for developing PTSD, rather than evolving as a consequence of the trauma (
27•).
In an mMRI study using a new scheme for cortical parcellation, Rauch et al. (
28) found selectively decreased rostral anterior cingulate and subcallosal cortical volumes in combat nurses with PTSD versus those without PTSD.
Functional imaging findings
Early functional studies in PTSD patients suggested abnormally elevated regional cerebral blood flow (rCBF) in the orbitofrontal cortex, insula, anterior cingulate cortex, and amygdala (
29,
30). Later, controlled investigations with PTSD have implicated many of the same areas that came to light in these early studies. In a neutral state study, Sachinvala et al. (
31) reported relatively greater rCBF in the bilateral anterior and posterior cingulate, the right temporal and parietal cortices, right basal ganglia, left hippocampus, and left orbitofrontal cortex. A limitation of this study is that the majority of the patients included were on psychotropic medications. When the five medication-free patients with PTSD only were compared with the healthy control group, increased rCBF was seen in the bilateral caudate and putamen, as well as the right orbitofrontal and bilateral anterior cingulate cortices. Semple et al. (
32) measured rCBF in a sample of patients with PTSD and comorbid substance abuse and a sample of healthy comparison subjects at rest and during an auditory continuous performance task. They reported that the PTSD group had greater rCBF in the right amygdala and left parahippocampal gyrus, and lower rCBF in the rostral anterior cingulate during these conditions.
Several studies of PTSD using symptom provocation paradigms have been reported that suggest group differences involving the amygdala, anterior cingulate, and areas of the prefrontal cortex. Using a variety of symptom provocation paradigms, several investigators reported increased amygdala activity in PTSD, compared with control individuals, in response to the symptom provocation condition (
33–
35). The majority of studies have reported a relative failure of patients with PTSD to recruit the anterior cingulate cortex compared with healthy control individuals in response to symptom provocation (
32,
36–
38,
39•), although this finding is not completely consistent (
34,
40). In order to look at the issue of anterior cingulate function in PTSD in greater depth, Shin et al. (
39•) designed a study to specifically test the functional integrity of the anterior cingulate cortex in patients with PTSD using an Emotional Counting Stroop task (
41), which is a reliable means of recruiting the anterior cingulate in healthy, nonpsychiatric individuals. The PTSD group failed to activate rostral anterior cingulate cortex in the combat versus general negative word conditions, whereas the non-PTSD group exhibited significant rostral anterior cingulate cortex activation. Similar results were later reported by Lanius et al. (
42) using fMRI and a high field strength (4 Tesla) magnet. Using a symptom provocation paradigm with scripted imagery to study patients with non–combat-related PTSD, brain activity was again lower in patients with PTSD compared with control individuals in the anterior cingulate and medial prefrontal cortex. In addition, reduced activity in the thalamus was thought related to disruption in normal intrathalamic sensory processing relays. Studies using symptom provocation paradigms have also demonstrated significantly greater activity in patients with PTSD compared with control individuals in brain regions associated with motor preparedness in response to threat, such as the amygdala, periaqueductal gray, primary and supplementary motor cortices, and the cerebellar vermis (
34).
Osuch et al. (
43) suggested that some of the differences in reported findings in symptom provocation studies in PTSD may be attributable to the presence or absence of flashbacks. They reported positive correlations between flashback intensity and left hippocampal, left inferior frontal, left somatosensory, and cerebellar cortices, brainstem, right insula, and right putamen. Flashback intensity was inversely correlated with bilateral superior frontal, fusiform, and medial temporal cortices. These results are consistent with several other studies showing relatively decreased superior frontal rCBF in patients with PTSD compared with control individuals in response to symptom provocation paradigms (
30,
38,
40). Lanius et al. (
44) examined the issue of whether patients with PTSD who respond to script-driven imagery with dissociative responses (without physiologic activation) differ in patterns of brain activation from patients with PTSD with reexperiencing or anxiety responses associated with physiologic activation. Their findings suggest that the pattern of brain activity associated with dissociation differs from that associated with reliving the traumatic event; dissociated individuals demonstrate greater activity in the right anterior cingulate, right medial prefrontal cortex, inferior and middle frontal gyri, and middle temporal gyrus, whereas patients with reexperiencing responses typically demonstrate decreased anterior cingulate and medial prefrontal activity (
36–
38).
Most recently, Clark et al. (
45) examined the pattern of brain activity in response to a trauma-neutral verbal working memory updating task. Compared with healthy control individuals, patients with PTSD demonstrated a lack of bilateral dorsolateral prefrontal cortex activation and a shift to bilateral superior parietal lobe activation. This finding, along with the earlier finding of decreased activation of Broca’s area (
30), suggests that PTSD may have a distinct pattern of brain activation in response to the organization and processing of verbal working memory. This is consistent with earlier proposals (
30,
37) that suggest a greater reliance in PTSD on nonverbal working memory and a shift away from verbal-based processing. Clark et al. (
45) suggested that this shift toward nonverbal processing is “important to the quality, nature, and intrusiveness of traumatic memories.”
In summary, functional neuroimaging findings in PTSD suggest that in threatening situations, patients with PTSD reallocate neural resources to more primitive limbic regions that mediate fear responding at the expense of cortical areas mediating higher cognitive functions. Evidence suggests that difficulties with verbal memory often observed in PTSD may be the result of a failure to recruit appropriate frontal temporal regions involved in verbal processing, with a shift toward more posterior brain regions (parietal lobes) typically involved in visuospatial processing and encoding. Overall, the pathogenesis of PTSD may be conceptualized as a fear conditioning process that is superimposed over some diathesis, which may involve a predisposition to amygdala hyperresponsivity, lack of sufficient functional connectivity between the anterior cingulate and the amygdala, hippocampal deficiency, and inadequate function of brain regions involved in executive function, as well as verbal memory and processing.