In the original report of association with schizophrenia in an Icelandic population, Stefansson and colleagues (
6) identified a “core at-risk haplotype” consisting of five SNPs (SNP8NRG221132, SNP8NRG221533, SNP8NRG241930, SNP8NRG243177, and SNP8NRG433E1006) and two microsatellites covering the 5′ end of the
NRG1 gene and extending into the second intron (hereafter referred to as the “deCODE haplotype”). Separate follow-up studies in Scottish, Irish, mixed United Kingdom, and Dutch populations confirmed the genetic association between schizophrenia and
NRG1 by using markers within the same core haplotype (
11–
14) or with overlapping markers in the 5′ region (
15,
16). Studies in four Asian populations also showed a strong association between schizophrenia and
NRG1 polymorphisms at the 5′ (
17–
20) and 3′ end of the gene (
19). Together these results, not withstanding two negative studies (
21,
22), provide strong evidence that
NRG1 is a schizophrenia-susceptibility gene. Additional support for NRG1’s role in schizophrenia comes from the phenotype of
NRG1 and ErbB4 mutant mice (
6,
23–
25), which exhibit behaviors similar to those of established rodent models of schizophrenia (
26).
To test for differential expression of
NRG1 in schizophrenia, we examined mRNA abundance for
NRG1 types I–IV in the human hippocampus, a region prominently implicated in the pathogenesis of schizophrenia (
30) and in the neurobiology of
NRG1 (
31). Examination of the effects of genetic variation on
NRG1 expression included 4 SNPs from the deCODE core haplotype (
6), with each SNP being tested individually for associations with
NRG1 mRNA levels in patients and controls. LD between SNPs was examined, and the deCODE at-risk haplotype region was tested for association with
NRG1 mRNA abundance. Our results support our primary hypotheses and indicate that the region of the gene implicated by the core at-risk haplotype impacts on specific
NRG1 isoforms and interacts with their expression in schizophrenia.
Discussion
We have investigated the expression of
NRG1 type I–IV mRNA in the human hippocampus and examined the effects of schizophrenia and disease-associated polymorphisms in the 5′ upstream region on expression of these transcripts. We hypothesized that the genetic association of
NRG1 with schizophrenia is mediated by altered expression of the gene based on the location and noncoding nature of the disease-associated polymorphisms and the fact that extensive sequencing of
NRG1 has failed to identify pathogenic coding mutations (
6). We report three principal findings: (i) up-regulation of type I expression in the hippocampus in schizophrenia, (ii) association of type I expression with a single SNP residing in the original deCODE risk haplotype, and (iii) association of type IV expression with a single SNP and a four-marker haplotype representing the 5′ upstream region of the original at-risk haplotype associated with schizophrenia. We provide evidence of association between disease linked-variation in
NRG1 and altered
NRG1 isoform expression in the brain, and we propose that altered transcript regulation is a potential molecular mechanism behind the genetic association of
NRG1 with schizophrenia.
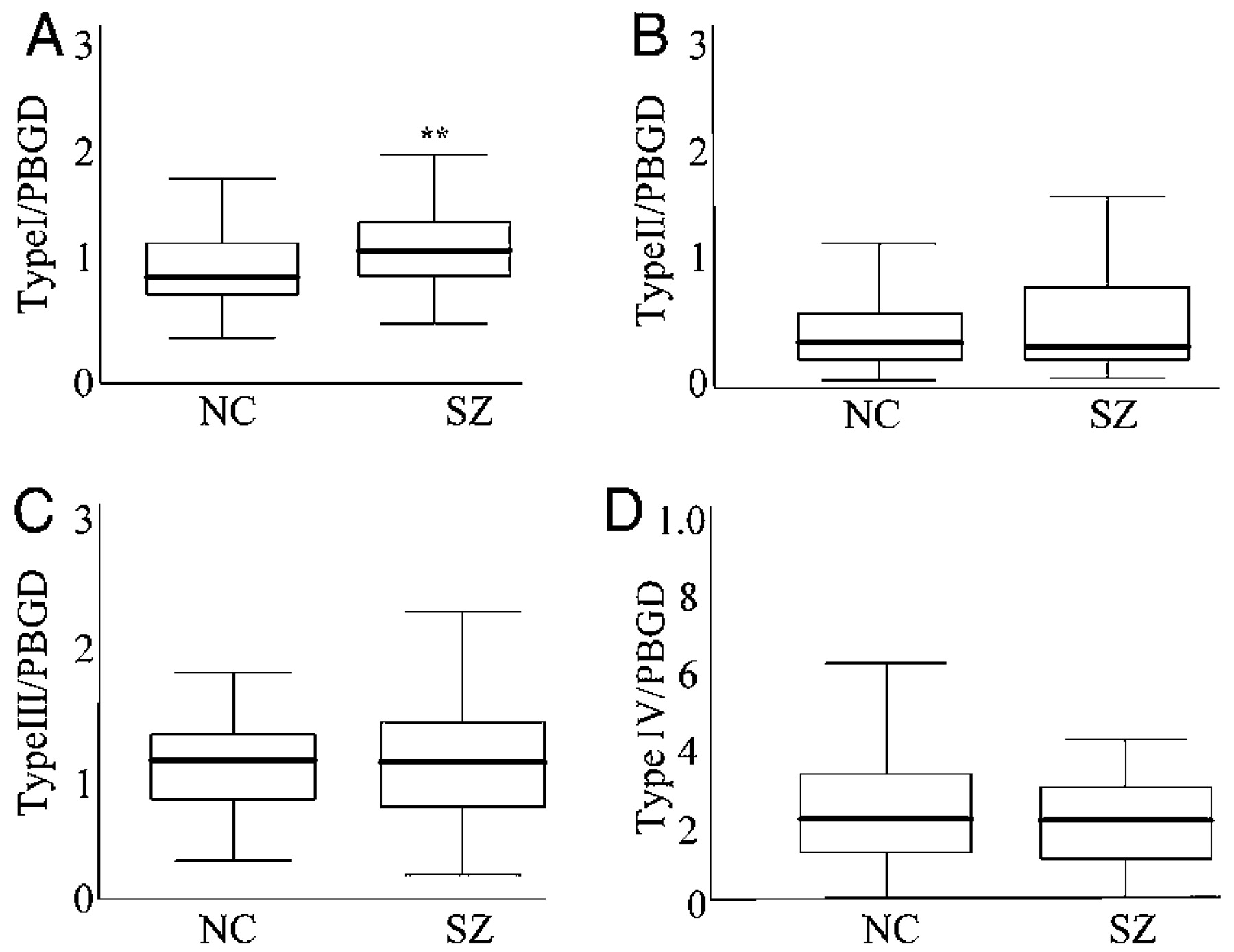
Our finding of increased type I mRNA
NRG1 expression in the hippocampus in schizophrenia replicates the finding in the dorsolateral prefrontal cortex of a smaller and separate brain series (
27). These findings suggest that enhanced type I expression is robust and found in two separate brain regions in schizophrenia. In addition, we also replicate the finding that type II and type III isoform expression is unaltered in schizophrenia, suggesting that these isoforms may not be directly relevant to the pathophysiology of the disease. However, we did observe increases in the relative abundance of type I to type II–IV, suggesting that the contribution of these latter isoforms to
NRG1 signaling in the hippocampus may be indirectly compromised in patients with schizophrenia. At present, it is unclear whether type I up-regulation in schizophrenia is primary or secondary to other abnormalities in
NRG1 isoform regulation or to other molecular changes associated with the disease.
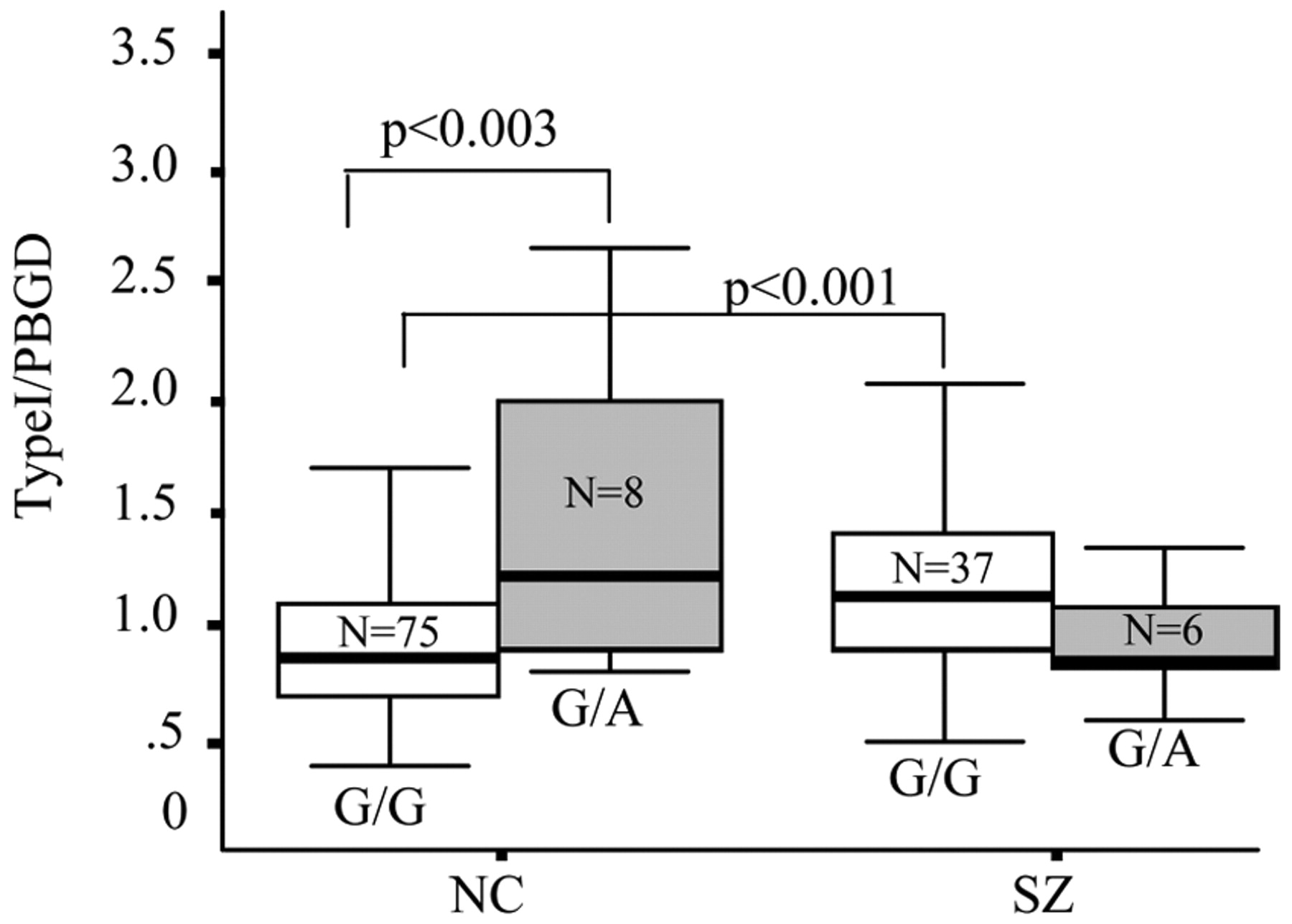
When the four individual SNPs representing the 5′ region of the deCODE at-risk haplotype were tested for association with type I
NRG1 mRNA, no main effects of genotype were seen for any of the SNPs. A diagnosis × genotype interaction was observed at a single SNP, SNP8NRG221132, and post hoc tests showed an effect of genotype only in control subjects on type I mRNA abundance. In a second independent cohort of brains in which increased type I mRNA expression previously had been reported in the schizophrenia samples, we found a main effect of a genotype at this SNP in the entire sample and again a genotype by diagnosis interaction. This main effect of the genotype was not seen in the first cohort; however, we note that the main effect in the entire sample is driven primarily by the controls. The observations that the main effect of the genotype was primarily in the controls, and that the four-marker risk haplotype had no effect on type I
NRG1 expression, raise the possibility that SNP8NRG221132 influences type I expression independent of its contribution to risk for schizophrenia. Additional support for the functional relevance of SNP8NRG221132 comes from the bioinformatic promoter analysis that predicts the risk allele (G) leads to a loss of binding for SRF. SRF is a transcription factor that regulates the expression of genes encoding cytoskeletal proteins, such as cofilin and actin (
32), both of which have been linked directly to
NRG1s role in actin dynamics (
33). The loss of SRF binding in controls homozygous for the risk allele, therefore, may be related to lower levels of type I mRNA transcription, as reported here. The direct functional consequences of this SNP for type I
NRG1 transcriptional control remain difficult to predict because the SNP resides 1 Mb upstream from the transcriptional start site of type I. However, SNP8NRG221132 could conceivably reside in a regulatory element of the gene, as is seen in other key developmental genes where genomic regions harboring cisregulatory elements can be located as far as 1 Mb from the transcription unit (
34).
The interrelationship between SNP8NRG221132, type I NRG1 expression, and schizophrenia is somewhat more difficult to interpret, because in contrast to the effect seen in controls, we did not see a similar genotype effect in the patients. This issue is discussed in Supporting Text, Discussion: Genetic Association and Type I Expression in Schizophrenia).
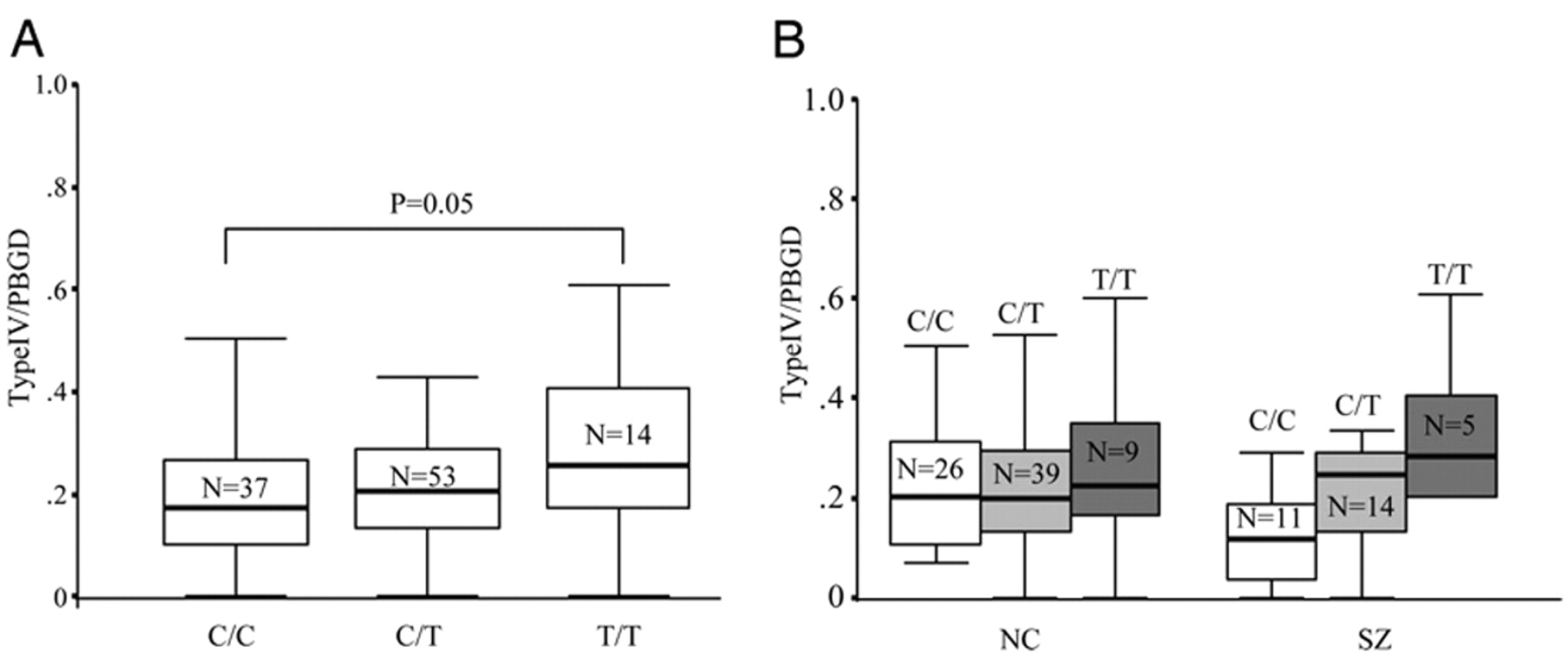
In contrast to the type I finding, which is not manifestly related to genetic variation in
NRG1 associated with schizophrenia, the association between both SNP8NRG243177 and the four-marker at-risk haplotype with expression of a novel isoform of
NRG1, type IV, suggests that we may have identified a genetic mechanism and a molecular phenotype underlying the involvement of
NRG1 in susceptibility for schizophrenia. The risk allele of SNP8NRG243177 and the deCODE haplotype predicted higher levels of type IV
NRG1 expression in our entire sample. Analysis of the three genotype groups for SNP8NRG243177 revealed that individuals homozygous for the risk allele had the highest levels of type IV expression, with evidence of an allele dose-dependant effect. This observation appeared more pronounced in the patients, although trends in the same direction were found in the normal controls and no diagnosis × genotype interaction was observed. SNP8NRG243177 is the most 3′ of the SNPs in the four-marker deCODE haplotype and is located ≈1.2 kb upstream of the transcriptional start of type IV. Because none of the other single SNPs in this haplotype were associated with type IV
NRG1 expression, our results suggest that SNP8NRG243177 is a functional polymorphic variant that regulates type IV
NRG1 mRNA levels or is in strong LD with a nearby functional mutation. Additional support for the functional relevance of SNP8NRG243177 for gene regulation comes from the bioinformatic prediction that this SNP determines a putative transcription factor binding domain for SRF, myelin transcription factor 1, and High Mobility Group Box Protein-1. Of note, SRF and myelin transcription factor 1 play critical roles in neuronal migration, synaptic plasticity, and oligodendrocyte proliferation and survival, respectively, providing a striking molecular convergence with current hypotheses regarding the neurobiology of schizophrenia and the potential role of
NRG1 (
35). However, we do not know which, if any, of these changes in transcription factor binding sites might mediate the association between SNP8NRG243177 and type IV expression and schizophrenia. Of potential interest is High Mobility Group Box Protein-1, an abundant chromatin-binding protein, which acts as an architectural facilitator in transcription (
36). In our sample, acquisition of two High Mobility Group Box Protein-1-binding motifs (i.e., homozygosity for the risk allele) was associated with significantly elevated type IV
NRG1 expression, whereas acquisition of one (i.e., heterozygosity for the risk allele) was not. This observation suggests (i) that this binding site may potentiate type IV
NRG1 transcription (and that SRF binding is necessary for optimal levels of type IV transcription) and 2) that this effect may be recessive.
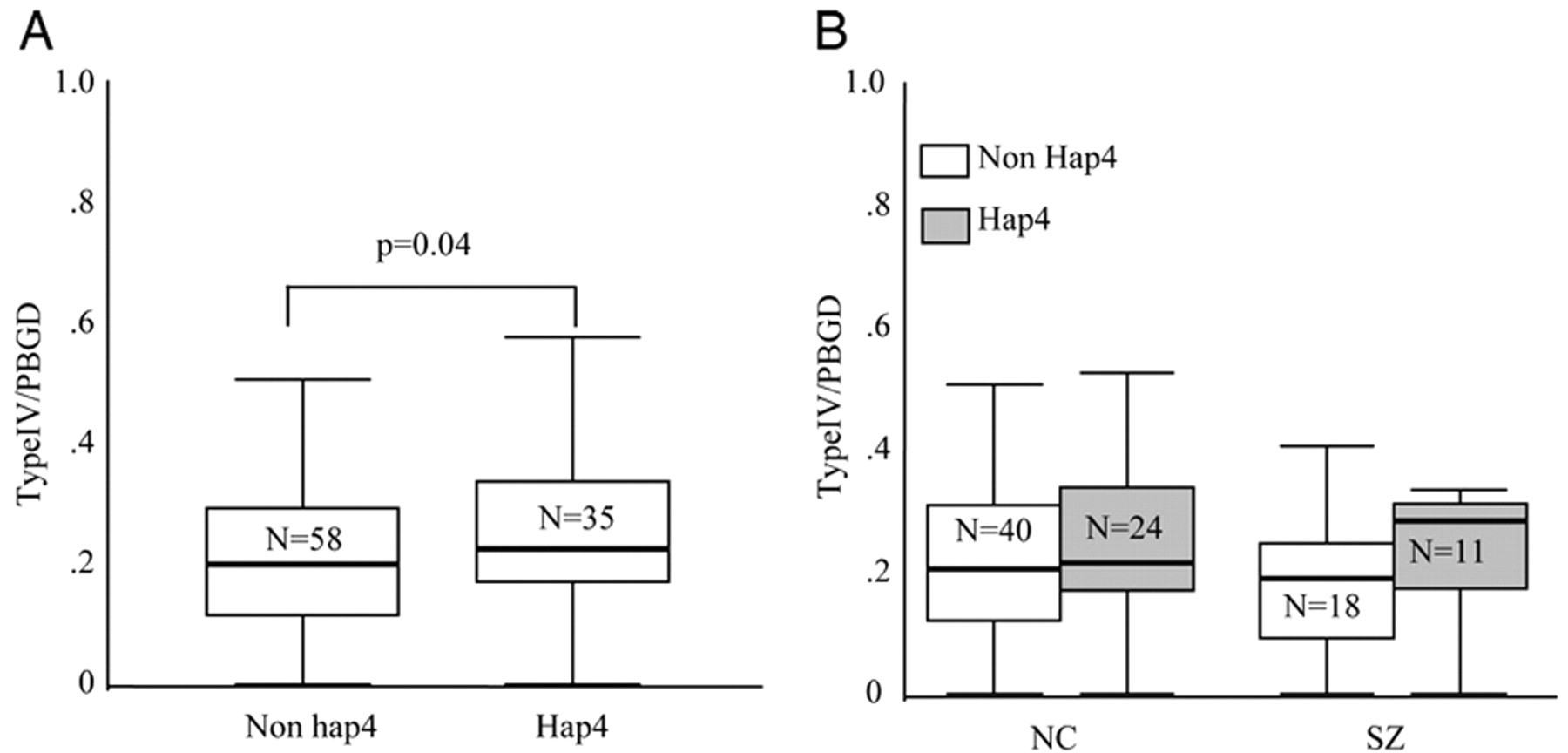
The absence of overall changes in type IV NRG1 gene expression levels in schizophrenia suggest that altered type IV, unlike type I, is not a general characteristic of the disease state, per se. Indeed, if altered NRG1 type IV expression is part of the genetic architecture of susceptibility for schizophrenia, it would not be expected to show an effect at the general population level, assuming that the at-risk haplotype is relevant for, at most, 10% of cases. Furthermore, our finding that the deCODE risk haplotype is associated specifically with type IV NRG1 expression argues that the clinical association with NRG1 is based on this molecular effect.
We further report association of type IV NRG1 mRNA in schizophrenia with five additional htSNPs, which span a 17-kb gap between the four SNPs from the deCODE haplotype. To our knowledge, these SNPs have not been tested for association with schizophrenia in the same clinical samples in which the deCODE SNPs were positive. We genotyped these SNPs to address the possibility that the deCODE haplotype might not provide sufficient information regarding genetic diversity in our sample. None of these SNPs showed main effects, and their association with NRG1 type IV expression in schizophrenia is likely via LD with SNP8NRG243177.
In our sample, the deCODE risk haplotype, which we termed hap4, was present in both Caucasian and African American populations but more common in the Caucasian sample. The significant degree of LD across this region of the gene suggests that, at least in Caucasians, it has undergone very little recombination (
37). Furthermore, the region is highly conserved between species, including chimpanzee, dog, mouse, and rat, suggesting that this region of the gene is functional, probably involved in transcriptional regulation of
NRG1 (
38). We found no evidence to suggest that the frequency of the deCODE haplotype was higher in our patient population compared with controls, but our sample is too small to meaningfully test for association with clinical phenotype. Of note, we observed that the frequency of hap2 was somewhat greater in the African American patients (34%) compared with African American controls (25%), suggesting that in different ethnic groups, different haplotypes in the same region of the gene may be associated with schizophrenia. However, because of the small sample size involved, conclusions are limited. Interestingly, hap2 in the African American sample contains the same allele at SNP8NRG243177 as hap4.
In the original report by Stefansson
et al. (
6), association in Icelandic families was mapped to a seven-marker haplotype spanning a 270-kb LD block starting at SNP8NRG221132 and ending with a synonymous SNP in exon two (SNP8NRG433E1006) and two microsatellites in the second intron (478N14-848 and 420M9-13950). Evidence of association to this region of the gene in other samples has been primarily to SNPs at the 5′ end of this haplotype, encompassing the SNPs typed in this study. Thus, although we cannot exclude the possibility that the causative mutation(s) accounting for our association with type IV lies downstream from our typed SNPs, we tend to doubt this possibility for three reasons: (i) the exon 2 SNP and the microsatellites typed in the deCODE haplotype have not shown single point (pairwise) association with schizophrenia in any study (
6,
18,
19,
39), in contrast to the four SNPs tested here, (ii) the physical location of SNP8NRG243177 (i.e., ≈1,200 bases upstream from the exon 1 start site) makes it a far better candidate for being located in a transcriptional regulatory region for type IV, and (ii) this SNP is in a putative functional transcription factor binding domain.
The known biological functions of
NRG1 (
9) fit well with current hypotheses regarding the neurobiology of schizophrenia (
35), including regulation of synaptogenesis,
in vivo synaptic transmission, long-term potentiation, activity-dependent synaptic plasticity, and neuronal migration as well as neurotransmitter function (NMDA, GABA, α-7, and dopamine) and oligodendrocyte biology, all of which are proposed to interact or be altered in schizophrenia (
30,
40). Of particular relevance is the recent finding that
NRG1 down-regulates NMDA-receptor currents in prefrontal cortical pyramidal neurons and slices (
41). These data suggest that increased expression of
NRG1 type I or IV would translate into decreased NMDA receptor-mediated signaling, one of the principal neurotransmitter hypotheses of schizophrenia.
Finally, it should be noted that we have performed a number of tests in this study, and correction for multiple testing was not performed. Correction for random effects, such as Bonferroni correction, would be an excessively conservative approach, particularly given that we have restricted our primary analyses to planned comparisons (based on strong prior clinical association and physical location of the SNPs) of four SNPs and a single haplotype comprised of these SNPs. Because the SNPs are in moderate LD, the degree of independence between markers is low and, therefore, correcting for multiple testing would result in a high type II error rate. The prior probability and the predictable association between the deCODE haplotype and expression of NRG1 isoforms (especially type IV, which is its immediate physical neighbor) combined with the LD between SNPs in this haplotype makes statistical correction for these comparisons inappropriate. Nevertheless, our finding regarding type IV expression and the deCODE haplotype and SNP8NRG243177 requires independent replication.
In summary, we provide evidence of splice variant-specific alterations of NRG1 gene expression in schizophrenia and demonstrate that disease-associated polymorphisms in a 5′ regulatory region of NRG1 are associated with differential NRG1 isoform expression. We suggest that the mechanism behind the clinical association of NRG1 with schizophrenia is altered transcriptional regulation, which modifies, probably to a small degree and in an isoform-limited fashion, the efficiency of NRG1 signaling effects on neural development and plasticity. Such alterations may compromise cortical and hippocampal function through one or more of the roles of NRG1 and reflect, at least partly, the contribution of NRG1 to the genetic risk architecture for the disease.