Major depression has become a health crisis of epidemic proportions in the modern world. The prevalence of major depression has risen over the last several generations in every country examined (
1), and age of symptom onset has decreased (
2). One in six individuals in the United States will experience an episode of major depression in his or her lifetime (
3), and the risk of subsequent episodes rises dramatically once a person has been depressed (
4). Between 10% and 15% of severely depressed people eventually commit suicide (
5), and studies indicate that depression significantly increases all-cause mortality (
6) and predicts the later development of a number of medical conditions, including cardiac and cerebrovascular disease (
7,
8), hypertension (
9,
10), diabetes (
11,
12), obesity and the metabolic syndrome (
13,
14), and cancer (
15).
Unfortunately, most patients with depression do not experience a complete resolution of symptoms with conventional antidepressant treatment, and 10%–20% of patients have depression that is refractory to all currently available modalities, including electroconvulsive therapy (
16). In addition to efficacy issues, many patients are unable to tolerate the side effects associated with antidepressants or electroconvulsive therapy. The risks of not responding to (or tolerating) treatment have been highlighted by recent studies documenting the fact that a partial—but incomplete—response is associated with an increased risk of full symptomatic relapse (even when the patient is receiving therapy) and worse long-term disease course, as well as significantly impaired quality of life (
17,
18). Treatment resistance also results in a six times increase in direct health care costs (
19). These factors highlight the tremendous need to identify novel treatment strategies for depression, especially for depressed patients whose depression is unresponsive to treatment or who are intolerant of conventional therapies.
THE IMMUNE SYSTEM AND DEPRESSION
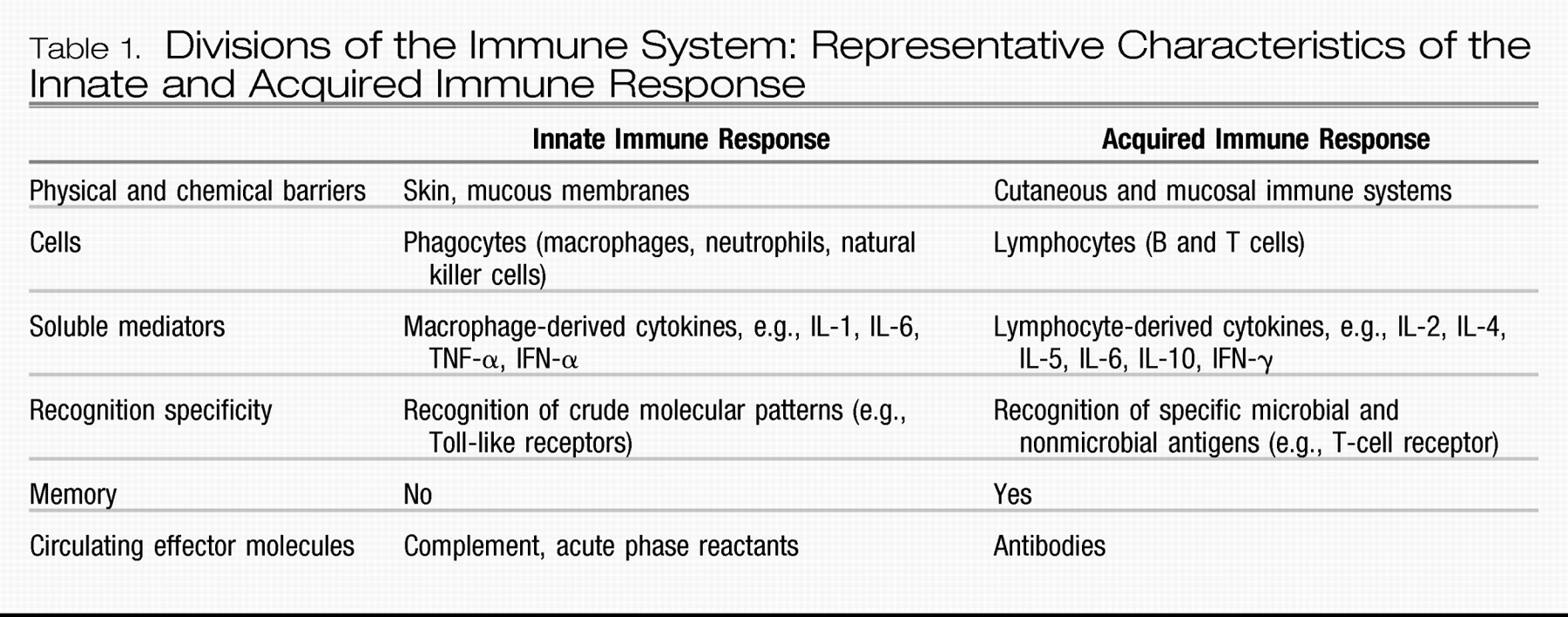
One consideration that has received increasing attention is the possibility that the immune system may contribute to the pathophysiology of depressive disorders and may thus represent a heretofore unrecognized and novel target for future research and therapeutic exploration. Although early studies focused largely on acquired immune responses in patients with depression (e.g., T- and B-cell functions, which were generally found to be suppressed), more recent research suggests that a significant percentage of depressed patients may experience activation of the innate immune response (
2021–
22).
In contrast to the acquired immune response (
Table 1), which develops slowly (i.e., over days) and is highly specific in its recognition of pathogens, the innate immune system provides a rapid, front-line defense against a variety of pathogens and damaged or dead cells, using relatively crude (nonspecific) pattern recognition receptors referred to as Toll-like receptors to initiate and mobilize the response to infection and/or tissue damage and destruction (
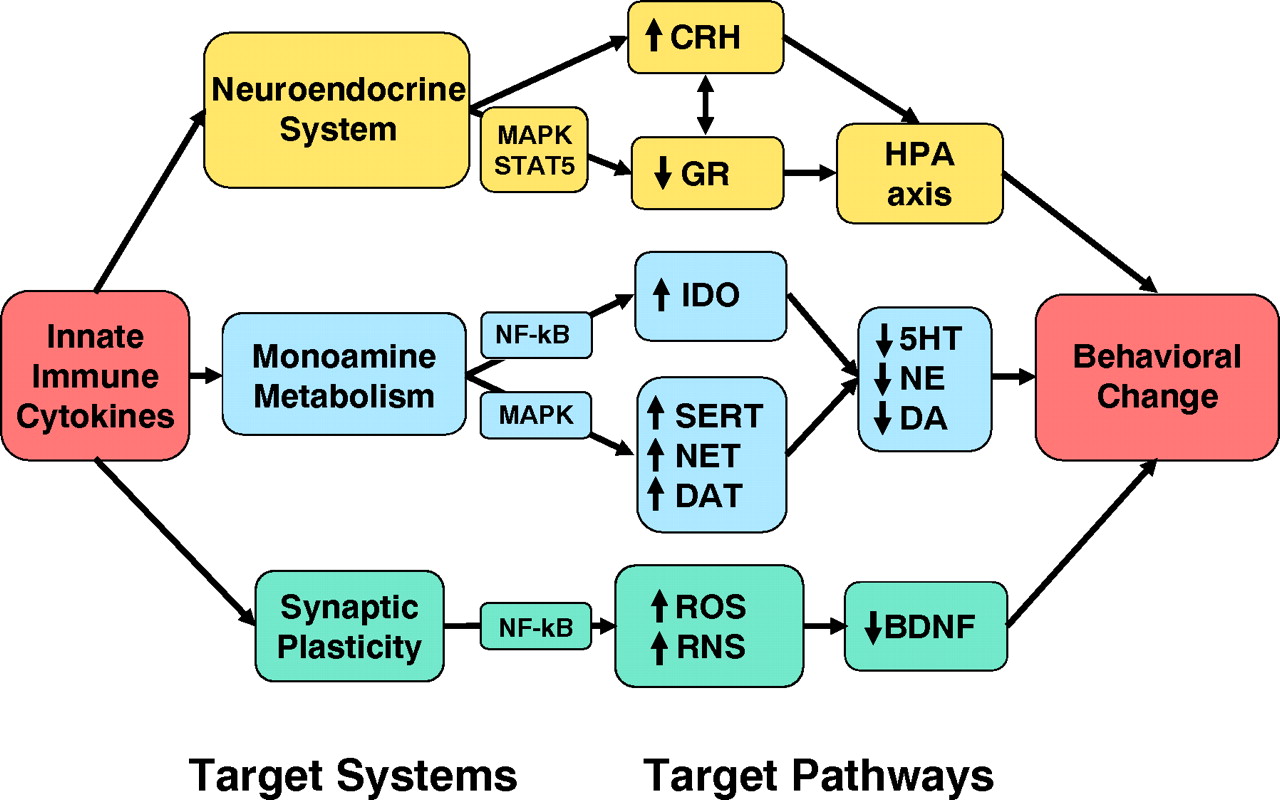
23). Toll-like receptors in turn are linked to fundamental inflammatory signaling pathways including nuclear factor-κB (NF-κB) and mitogen-activated protein kinases (MAPKs), which when activated stimulate the production of the innate immune cytokines interferon (IFN)-α, interleukin (IL)-1, IL-6, and tumor necrosis factor-α (TNF-α); chemokines; adhesion molecules; and other inflammatory mediators including the prostaglandins, histamine, and reactive oxygen and nitrogen species (
23,
24). These molecules then orchestrate the local immune response by recruiting and activating relevant immune cells, which leads to the swelling (tumor), redness (rubor), heat (calor), and pain (dolor) that constitute the clinical characteristics of inflammation. Innate immune cytokines also enter the peripheral blood and stimulate local nerve fibers (see below) to mobilize a systemic response to infection and tissue trauma that includes activation of the acute-phase response in the liver, which involves the production of acute-phase proteins such as C-reactive protein (CRP), and a central nervous system (CNS) response, which involves fever, fatigue, reduced environmental exploration, anorexia, and altered sleep. This CNS response, which has been referred to as “sickness behavior,” is believed to represent a reorganization of behavioral priorities to conserve and divert essential energy resources to pathogen elimination, tissue repair processes, and protection from future injury or attack (
20,
22,
25).
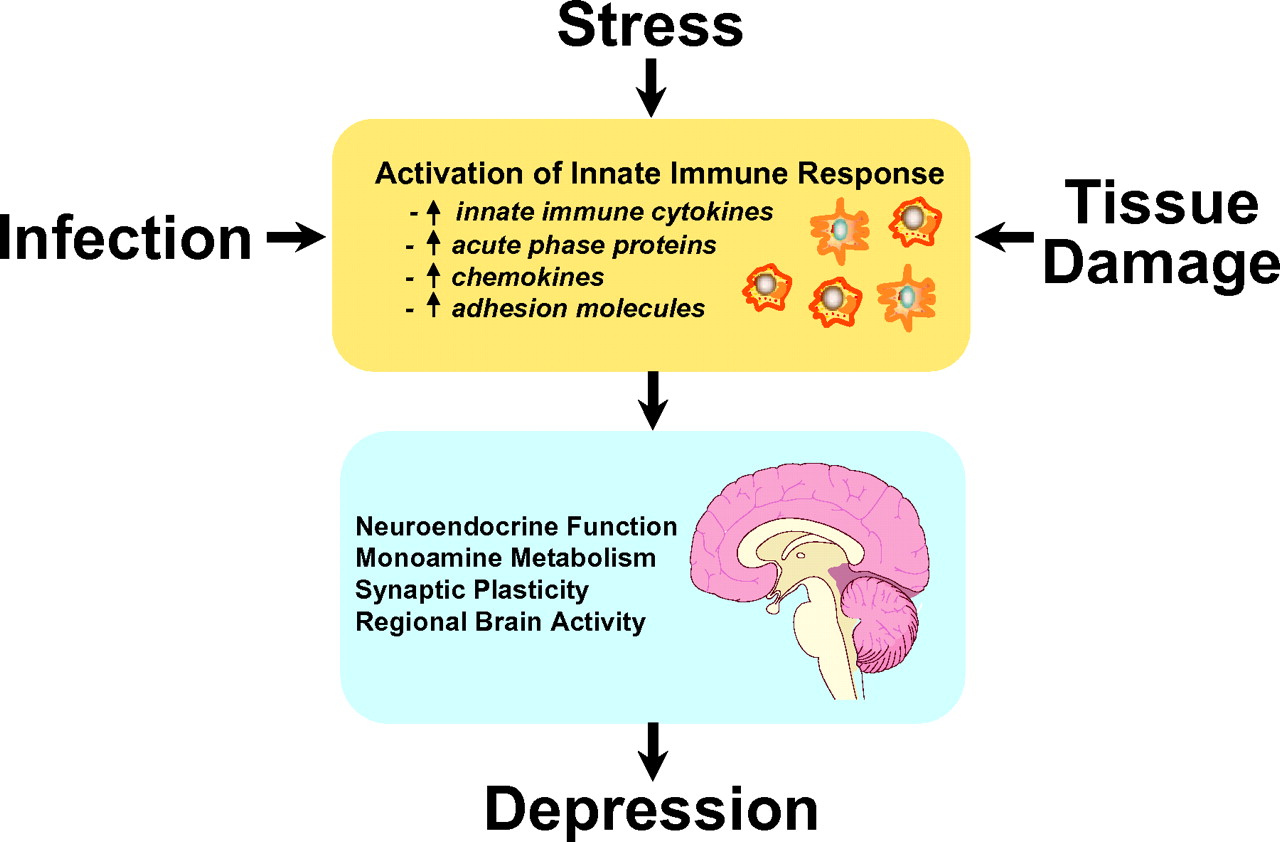
A major breakthrough in terms of the recognition of the potential contribution of the immune system to depression has been the demonstration that all of the cardinal features of inflammation are apparent in patients with major depression (
Figure 1). Patients with major depression have been found to exhibit significant elevations of innate immune cytokines and their soluble receptors in both peripheral blood and cerebrospinal fluid (CSF) and have exhibited elevations in acute-phase proteins, chemokines, and adhesion molecules as well as inflammatory mediators such as the prostaglandins. Given the number and variety of studies done in this area, a meta-analysis has been conducted, and the data indicate that of these markers of inflammation, elevations in IL-6 and CRP are some the most reliable (
26). Because of the relationship between body mass index and CRP and IL-6 (adipocytes are capable of producing IL-6 as are macrophages within fatty tissues) (
27), elevations of these markers in patients with obesity should be interpreted with caution. In addition to mean increases in inflammatory biomarkers in depressed patients versus control subjects, correlations between depressive symptom severity and increases in measures of peripheral inflammation have been observed in multiple studies (
20,
28–
31). Although the linkage of inflammatory markers with specific behavioral profiles is still under investigation, it should be noted that fatigue, loss of energy, and psychomotor retardation are some of the most common symptoms after cytokine administration (
32,
33).
Another major body of evidence supporting an immune system contribution to the development of depression is the profound behavioral disturbances that occur in patients treated with the innate immune cytokine, IFN-α. IFN-α has both antiviral and antiproliferative activities and is therefore used to treat both infectious diseases and cancer (
34). Although an effective therapy, IFN-α is notorious for causing a variety of behavioral alterations including symptoms sufficient to meet criteria for major depression in up to 50% of subjects, depending on the dose (
34,
35). Treatment of patients before and during IFN-α therapy with antidepressants has been shown to markedly reduce the incidence of depression (
35–
37), supporting the notion that cytokine-induced depression not only shares behavioral similarities with major depression in otherwise healthy individuals but also shares pharmacological response characteristics. Of note, rhesus monkeys administered IFN-α also exhibit depressive-like huddling behavior that was initially described in monkeys administered the monoamine-depleting drug, reserpine (
38).
A final consideration regarding the role of the immune system in depression is the high rate of depression in medically ill patients with disorders that involve the immune system including infectious diseases, cancer, and autoimmune disorders (
39). Rates of depression are on average 5–10 times higher in these diseases (
40), and studies have shown a relationship between inflammatory markers and depression and other behavioral alterations in these disorders (
41–
45). These data indicate that there appears to be a specific relationship between inflammation and behavioral symptoms as opposed to a more nonspecific relationship between being ill and emotional distress. Further supporting the specificity of the cytokine-depression link in those who are medically ill is that patients with autoimmune disorders treated with cytokine antagonists have exhibited significant improvement in depressive symptoms (see below) (
46,
47). In addition, there is increasing recognition that inflammation may play a prominent role in a number of common disorders including cardiovascular disease, diabetes, and metabolic syndrome and cancer—all disorders with increased rates of depression (
48–
50). Taken together these data suggest that inflammation may be a shared pathology between these diseases and depression.
STRESS, DEPRESSION, AND THE INNATE IMMUNE RESPONSE
One of the most profound discoveries that has linked depression and the immune system is the finding that psychosocial stress, a well-known precipitant of mood disorders, can activate the innate immune response. For example, in a study of normal volunteers subjected to the Trier Social Stress Test (a public speaking task followed by mental arithmetic), examination of peripheral blood inflammatory markers revealed significant increases in NF-κB DNA binding within minutes after stressor cessation (
84). These data complement results from a host of studies demonstrating that a variety of both acute and chronic emotional and physical stressors are associated with increases in inflammatory markers including innate immune cytokines and their soluble receptors as well as acute-phase proteins (
85–
87). Interestingly, the innate immune response to stress appears to be exaggerated in depressed patients exposed to early life stress (ELS). Indeed, depressed male patients with increased ELS exhibited significantly greater peripheral blood IL-6 responses and increased NF-κB DNA binding in response to the Trier Social Stress Test than nondepressed control subjects (
88). This relationship between ELS and increased inflammation has also been observed in a large population-based study wherein individuals exposed to increasing levels of ELS were found to exhibit increasing levels of CRP in adulthood (
87). Given the relationship between stress, depression, and inflammation, these data raise the question as to whether inflammation plays a role in the link between stress, depression, and disease, especially given the recent recognition that inflammation may represent a common mechanism for a number of illnesses including cardiovascular disease, diabetes, and cancer (
48–
50,
89,
90). As noted above, the effects of stress on relevant growth factors through its effects on the immune system may also play a role in neurodegenerative disorders (in addition to depression) (
91).
Regarding the mechanism(s) by which stress influences the innate immune response, emerging data from humans and laboratory animals indicate that the sympathetic and parasympathetic nervous systems may be involved. Antagonism of both α- and β-adrenergic receptors has been shown to abrogate the effects of stress on the induction of innate immune cytokines in both the peripheral blood and brain of laboratory animals (
92). Interestingly, increased peripheral blood IL-6 concentrations due to the stress of increased altitude were eliminated by administration of the α-adrenergic agent, prazosin (
93). It should be noted, however, that β-agonists are known to have potent anti-inflammatory effects (
94,
95), and therefore the role of catecholamines in the regulation of the innate immune response is in need of further study. Indeed, there is some suggestion that stress or inflammation-related induction of α
1-adrenergic receptors (in combination with the effects of stress or inflammation on the β-receptor) may be an important component in determining the net effect of catecholamines on the inflammatory response (
96). Finally, there has been recent interest in the role of parasympathetic pathways in inhibiting inflammation. For example, stimulation of the vagus nerve (and parasympathetic outflow pathways) has been shown to inhibit endotoxin induction of TNF-α and the signs of sepsis (
97). These effects appear to be mediated by the release of acetylcholine, which through binding to the α7 subunit of the nicotinic acetylcholine receptor can inhibit NF-κB. Nevertheless, given the rich interconnection between sympathetic and parasympathetic nervous systems including shared mediators, the exact mechanism by which the autonomic nervous system modulates the inflammatory response is an area that warrants further study.
THERAPEUTIC IMPLICATIONS
Given the potential role of the immune system in the development of depression, there has been mounting interest in targeting the innate immune response as a novel therapeutic strategy to treat depression. Of relevance in this regard, successful antidepressant treatment of major depression with selective serotonin reuptake inhibitors or tricyclic antidepressants has been associated with reduced circulating cytokine concentrations, including TNF-α (
98,
99) and IL-6 (
100) Recent observations suggest that bupropion, a marketed antidepressant, can also reduce circulating concentrations of TNF-α in mice and in subjects with inflammatory disorders including Crohn's disease (
101,
102). In vitro studies also indicate that a number of antidepressants can suppress the release of inflammatory cytokines while increasing the release of cytokines that inhibit inflammation such as IL-10 (
103). Importantly, patients with increased plasma concentrations of innate immune cytokines are less likely to respond to currently available therapies (
100,
104–
107) and, conversely, patients with treatment-resistant depression have been shown to be especially likely to demonstrate evidence of an activated innate immune response, including elevated plasma concentrations of innate immune cytokines (
100,
104–
108). These findings suggest that pharmacological strategies aimed to downregulate inflammatory signaling pathways may have unique antidepressant efficacy and may be especially relevant in patients with treatment-resistant depression with increased inflammation.
Preliminary data suggest that targeting the innate immune response may be a viable antidepressant strategy. For example, increased response rates and improvement of depressive symptoms in patients with major depression was reported in an add-on study using the anti-inflammatory agent, celecoxib, a cyclooxygenase-2 inhibitor, in combination with reboxetine (
109). In addition, in patients with psoriasis, the anti-TNF-α agent, etanercept, was found to reduce symptoms of depression (determined by the Hamilton Depression Rating Scale and the Beck Depression Inventory) independently from the clinical improvement of the primary disorder (
46). Animal data are also consistent with the notion that immune targeted therapies may have antidepressant potential. Indeed, mice who have had the gene for the TNF-α receptor “knocked out” have been found to exhibit an antidepressant phenotype and are resistant to the anxiety-inducing effects of viral infection (
110,
111).
Taken together, the data suggest that interventions that block inflammation may hold therapeutic promise especially in depressed individuals with increased inflammation. Given established cutoff values for increased inflammation as defined by the American Heart Association (i.e., CRP >3 ng/liter) (
48), such patients may be readily identified for further study and exploration of novel treatment approaches targeting cytokines or their signaling pathways.