Once a genetic basis for a condition has been demonstrated by genetic epidemiological studies, molecular genetic studies are undertaken to attempt to identify the particular genes underlying that basis. As explained in greater detail by other articles in this issue, the two main types of molecular genetic approaches are linkage and association studies, as applied to MDD below. Before these are reviewed in detail, they need to be placed in their historical context. In classic Mendelian genetic disorders that display specific patterns of familial transmission such as autosomal dominant or recessive, a relatively rare genetic disease strongly aggregates in certain families due to relatively large detrimental effects of a chromosomal abnormality or changes in a specific gene that changes its functional integrity. However, MDD, with complex patterns of genetic transmission similar to those of other common medical conditions such as hypertension or type 2 diabetes, is probably due to the accumulated effects of many genes of small, more subtle effects that interact with each other and environmental factors longitudinally across one's lifespan to “cause” an individual to develop the condition. Many of the early studies, not appreciating this distinction, did not recruit large enough numbers of subjects, making them insufficiently powered to detect genes of small effect. This limitation applies to both linkage and genetic association studies of psychiatric disorders such as MDD, providing one reason among several to explain why it has been difficult to unambiguously identify the liability genes for these conditions.
Association studies
Genetic association studies, which may take the form of case-control comparisons in unrelated individuals or family-based transmission tests, allow one to test specific genes or markers within genes for their contribution to normal traits or illness. Most association studies have thus far involved testing one or a few candidate genes implicated indirectly from other data, such as pharmacological agents (e.g., the gene for the serotonin transporter, which underlies the mechanism of action of selective serotonin reuptake inhibitor antidepressants), stress-related biology (e.g., hypothalamic-pituitary-adrenal axis genes), or animal models of depression (e.g., serotonin 1A receptor gene knockouts). The most popularly studied polymorphism in this respect is the 44-base pair insertion/deletion polymorphism (5-HTTLPR) occurring in the promoter region of the serotonin transporter gene (
SLC6A4). Both positive and negative reports abound for relating this genetic variant to a myriad of depressive-related phenotypes, making it difficult to draw solid conclusions about its etiological role. The most recent meta-analyses in MDD found a modest main effect for the short (S) allele of this polymorphism (
30,
31). Meta-analyses of related phenotypes suggest that there are small but statistically significant effects in association with neuroticism (
32–
34), suicide (
35–
37), and obsessive-compulsive disorder (
38) but not panic disorder (
39). In addition, there is mixed evidence for and subsequent debate over gene-environment interaction effects between 5-HTTLPR and stressful life events (
40–
42). Such an interaction implies that variation in this gene may increase liability to MDD primarily in individuals who experience significant lifetime stressors such as maltreatment or loss [“genetic control of sensitivity to the environment” (
43)].
Similar to linkage studies, most genetic association studies of MDD have been insufficiently powered because of relatively small sample sizes, resulting in a poor record of replicable findings similar to other complex traits (
44). Meta-analyses attempt to overcome this limitation by pooling data across studies, effectively increasing the total sample analyzed. However, they are often limited by the inability to include data from all available investigations owing to issues such as heterogeneity between studies or other criteria designed to maximize the quality of the results. Besides the serotonin transporter, separate meta-analyses have examined association of MDD with the gene encoding the dopamine D4 receptor (
DRD4) (
45), methylenetetrahydrofolate reductase (
MTHFR) (
46), the serotonin 2A receptor (
HTR2A) (
47), tyrosine hydroxylase (
TH) (
48), and angiotensin-converting enzyme (
ACE) (
49), with only the first two showing significant association across included studies. A recent study reviewed the extant literature for all candidate gene association studies of MDD published before June 2007 (
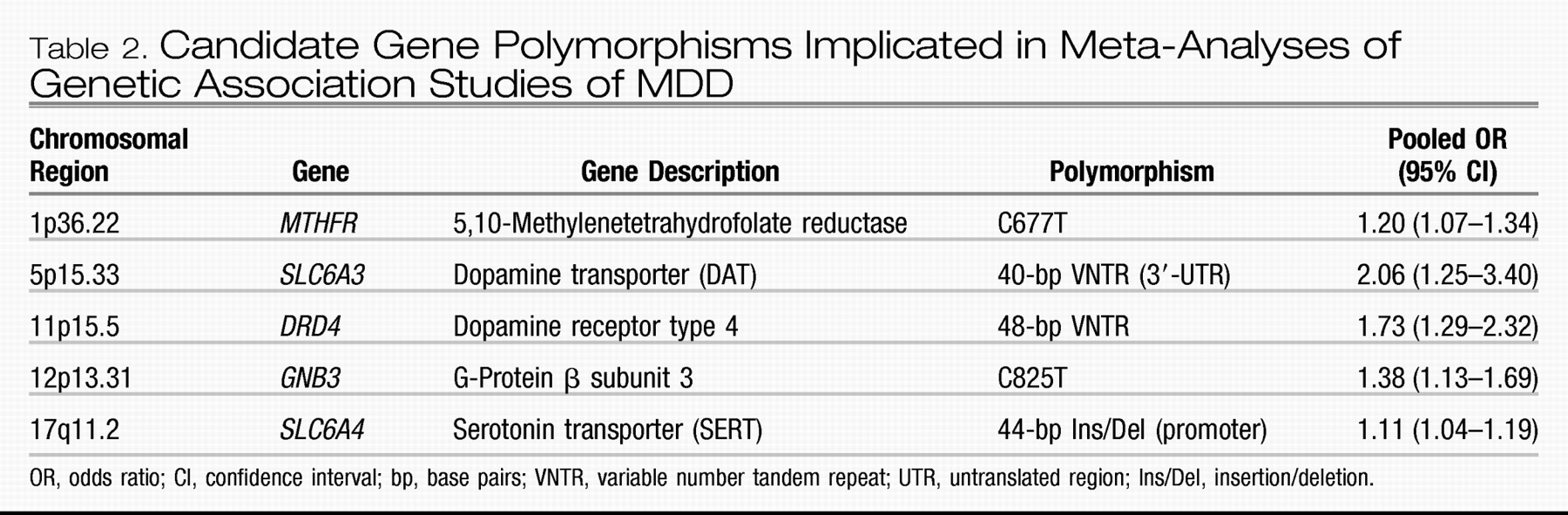
30). This study identified 183 articles that analyzed 393 polymorphisms in 102 candidate genes specifically for association with MDD using acceptable genetic methodology. The authors performed new meta-analyses for 20 polymorphisms in 18 genes that had available data from three or more studies not included in prior meta-analyses. Based on their new analyses together with the prior ones, there is statistical evidence supporting some role for at least five genes in liability to MDD (listed in
Table 2):
MTHFR,
SLC6A3,
DRD4,
GNB3, and
SLC6A4. [Note: Since this analysis, a recent, large study failed to find association with
MTHFR (
50).] There are probably many more genes involved in MDD, but either (1) insufficient evidence exists for those already studied because of too few or inadequately powered studies or (2) they have yet not been identified as probable candidates for study because of our limited understanding of the pathophysiology of MDD.
To overcome the latter limitation, complex genetic conditions are now being studied with genome-wide association studies (GWASs) that use breakthroughs in high-throughput genotyping technology to interrogate upward of hundreds of thousands of polymorphisms across the genome simultaneously in one experiment rather than gene by gene as is done in candidate gene studies. Three GWASs of MDD have been published to date and one is in press. The first, by Muglia et al. (
51), reports the findings of GWASs in two large, independent Caucasian samples: (1) 1,022 patients with recurrent MDD and 1,000 matched control subjects recruited from clinical settings in Munich, Germany, and (2) 492 subjects with recurrent MDD and 1,052 healthy control subjects recruited from a community survey in Lausanne, Switzerland. The researchers analyzed about 522,000 single nucleotide polymorphism (SNP) markers in the first sample and about 370,000 in the second. Although the analyses, separately or combined via meta-analysis, did not provide any findings meeting formal genome-wide statistical significance, the authors provided a table of 27 SNPs on 14 chromosomes that provided nominal but consistent evidence for association to MDD across the two samples. Of note, all of these are novel findings; i.e., they are in or near genes that have never been examined before in relation to MDD. The authors also examined association for candidate genes that have been tested in prior mood disorder studies;
GRM7, the gene for the metabotrophic glutamate receptor 7 implicated in a recent GWAS of bipolar disorder, emerged as the most significant of these.
Sullivan et al. (
52) conducted another large GWAS using the Genetic Association Information Network-MDD (GAIN-MDD) sample: 1,738 Dutch subjects with MDD recruited from clinical and community settings and 1,802 control subjects selected primarily from the Netherlands Twin Registry as having low genetic liability for depressive or anxiety disorders. Similar to the first study, this one also did not identify any associations with genome-wide significance after testing of approximately 435,000 SNPs, and their best signals were from novel candidates, although with no substantial overlap with the top candidates as reported in the first study. They followed up the most convincing candidate gene,
PCLO on chromosome 7 coding for the presynaptic protein Piccolo, by testing its association to MDD in five independent samples totaling 6,079 individuals with MDD and 5,893 control subjects. In only one of the replication samples did several of the
PCLO SNPs show modest association, although, overall, strict guidelines for replication were not met. However, in a reanalysis of those data Bochdanovits et al. (
53) claimed that there is support for a role in liability to MDD for a nonsynonymous coding SNP (rs2522833) in
PCLO. Other replication attempts are currently in progress. An independent study performed in a population-based cohort of elderly Dutch individuals recently reported association of rs2522833 with depressive disorders (
54).
The third GWAS for MDD (
55) was performed in 1,221 Caucasian patients selected from those who participated in the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) (
56), a multisite clinical trial; the 1,636 control subjects were drawn from subjects who screened negative for MDD, bipolar disorder, or schizophrenia from the Molecular Genetics of Schizophrenia study in GAIN (
57). That analysis, like the others, did not identify any markers with genome-wide significance. Their most strongly associated signals came from a region containing no known genes on 19q12. In that article, the authors combined their data with those from two other MDD GWASs: the Sullivan et al. study in the GAIN-MDD sample described above and an analysis performed in the same 1,636 control subjects from the Molecular Genetics of Schizophrenia sample together with 1,020 subjects from the Genetics of Early-Onset Major Depression (GenRED) sample (listed in
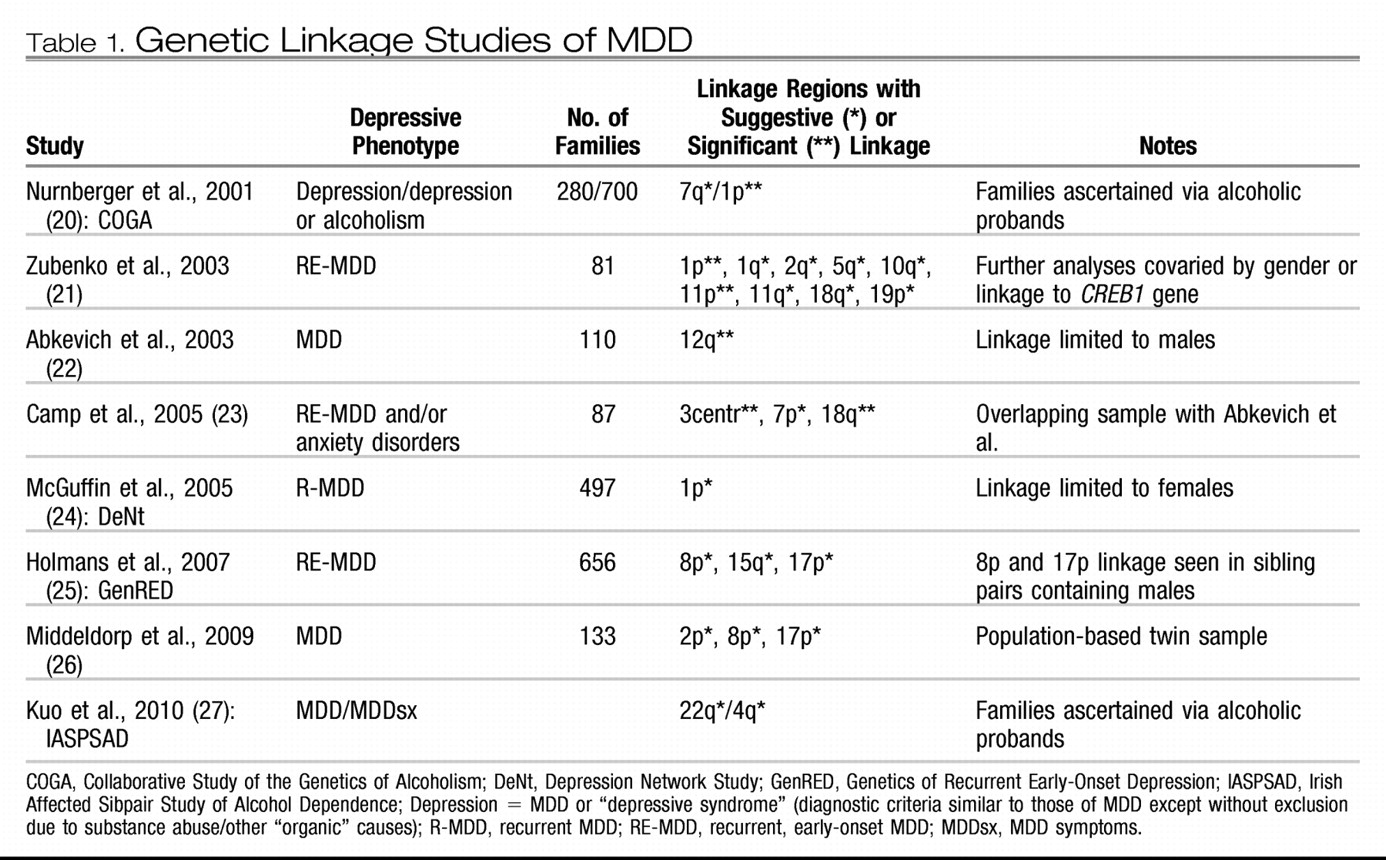
Table 1 under linkage analyses) and its follow-up, GenRED II (
58). They performed a meta-analysis of these three studies using more than 2.4 million imputed (estimated) SNPs, the most promising of which implicated three genes: (1)
ATP6V1B2 on 8p21.3, a gene also modestly implicated in a bipolar disorder GWAS that encodes for a vacuolar proton ATPase, (2)
SP4 on 7p15.3, encoding a brain-specific zinc-finger transcription factor, and (3)
GRM7 on 3p26.1, the same metabotrophic glutamate receptor gene mentioned in the first MDD GWAS above.