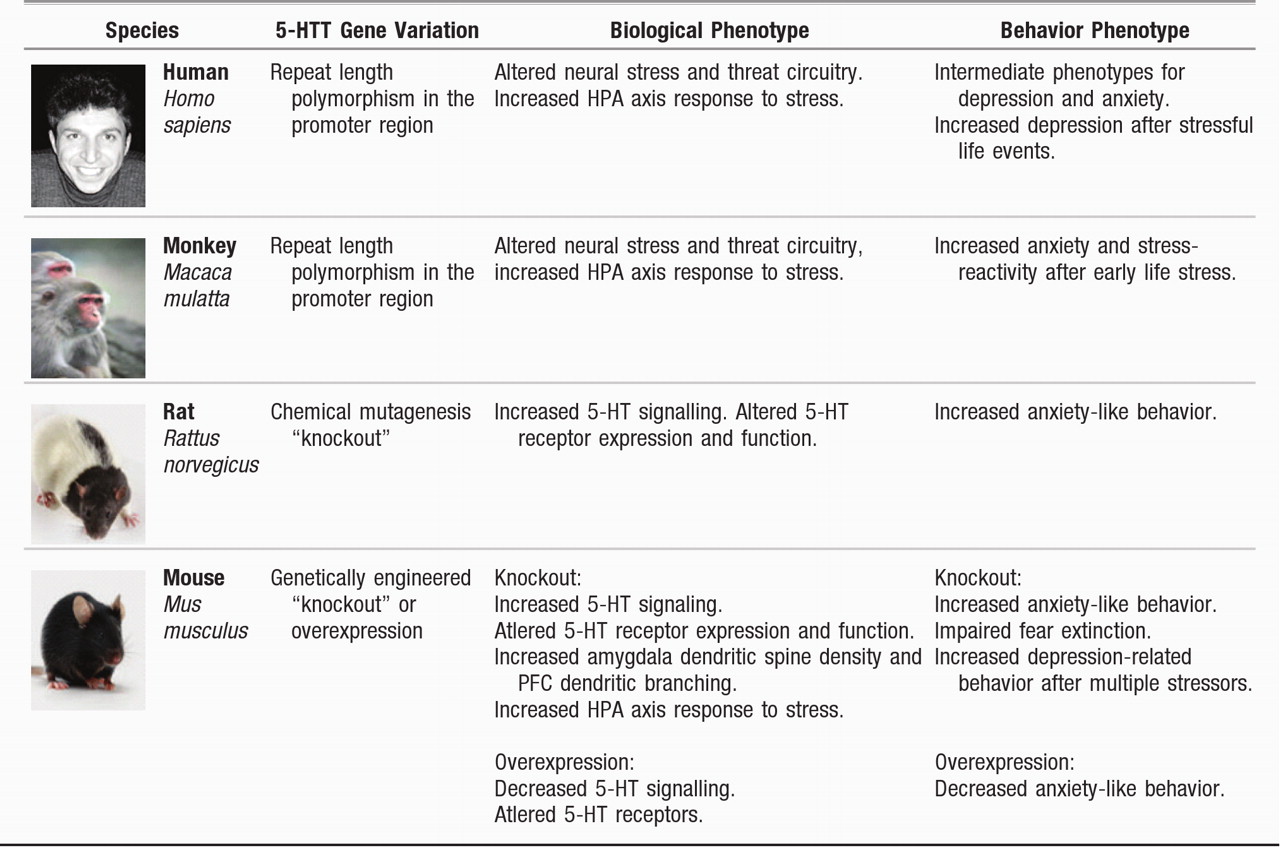
It is evident from research conducted with multiple species and from research using both observational and experimental methods that variation in
5-HTT modifies organisms' stress responses to their environments (
Figure 1). Complementary experimental and observational research designs are integral to testing not only the
5-HTT stress-sensitivity hypothesis, but all GxE hypotheses (
4,
5). Experiments with humans, nonhuman primates, and rodents elucidate biological mechanisms behind the hypothesis and also validate findings from human observational studies by using designs with stronger internal validity (e.g., by random assignment to stress conditions). Observational studies use designs with stronger external validity (e.g., by studying real-world stressors), estimate the effect size of the
5-HTTLPR GxE interaction in the human population, and allow researchers to study clinical depression as the outcome.
Human observational studies
The initial GxE effect (
3) did not have an overwhelmingly impressive p value, but it was robust, having been 1) discovered in an epidemiologically sound longitudinal cohort study; 2) tested in a straightforward and transparent analysis; 3) reproduced across two stressors, child maltreatment and adult stressful life events; and 4) reproduced across four depression phenotypes. How has this hypothesis fared in observational studies since it was initially tested?
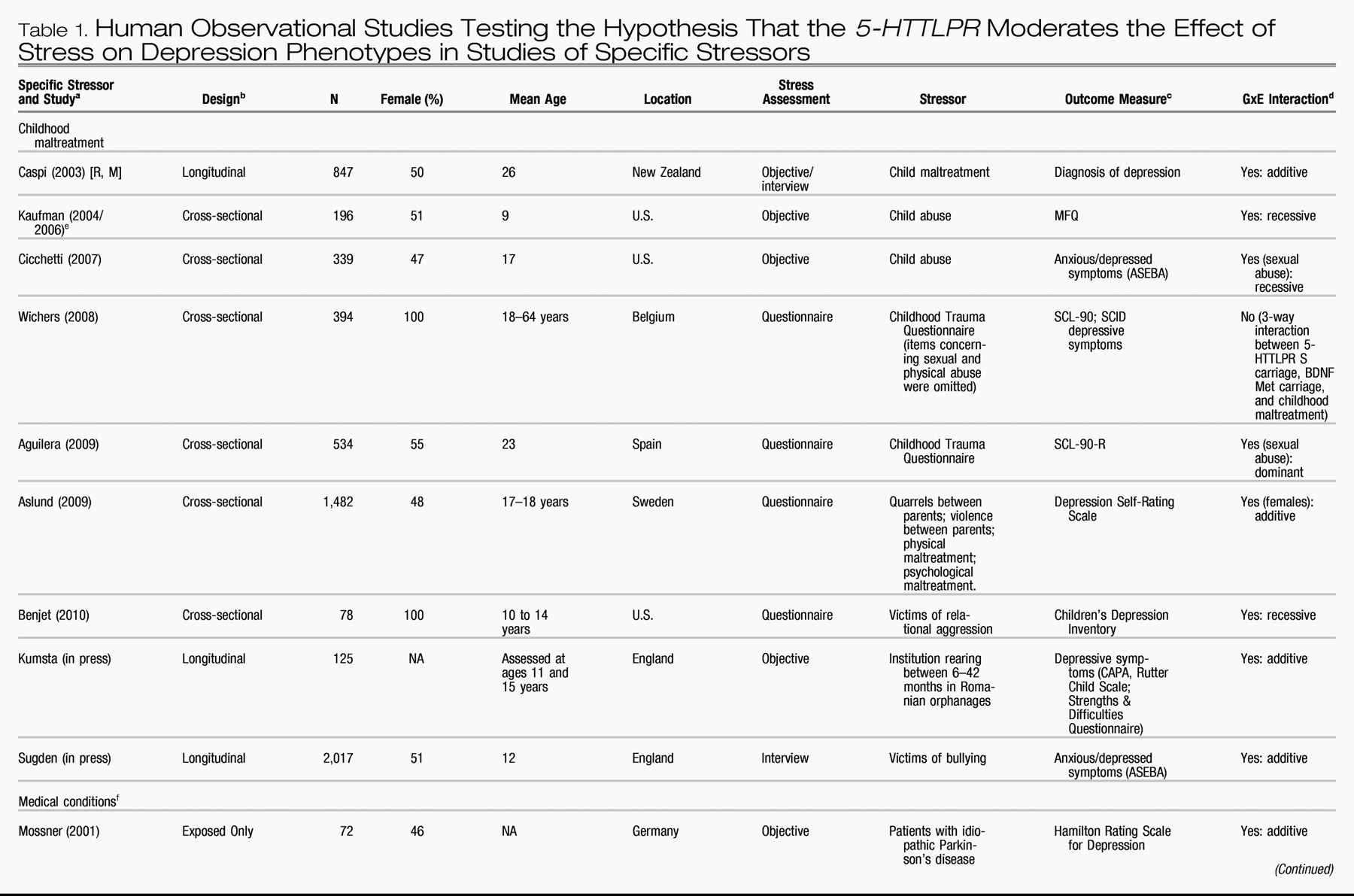
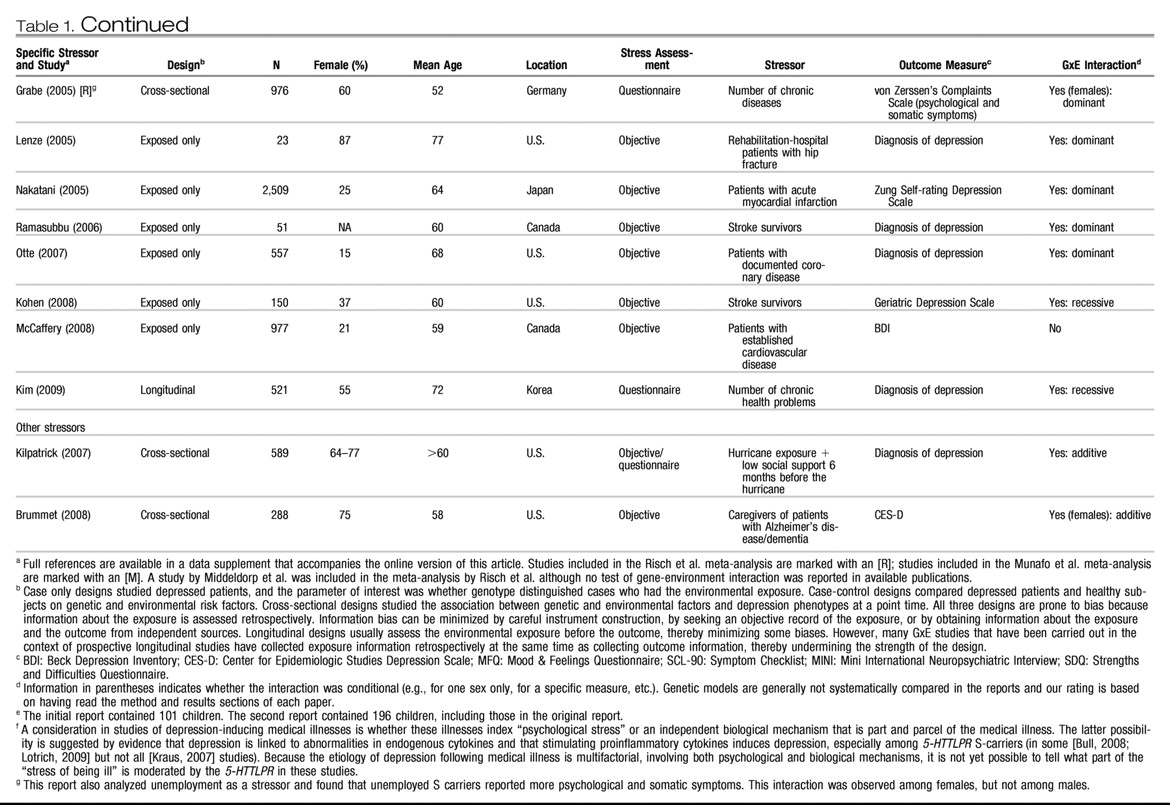
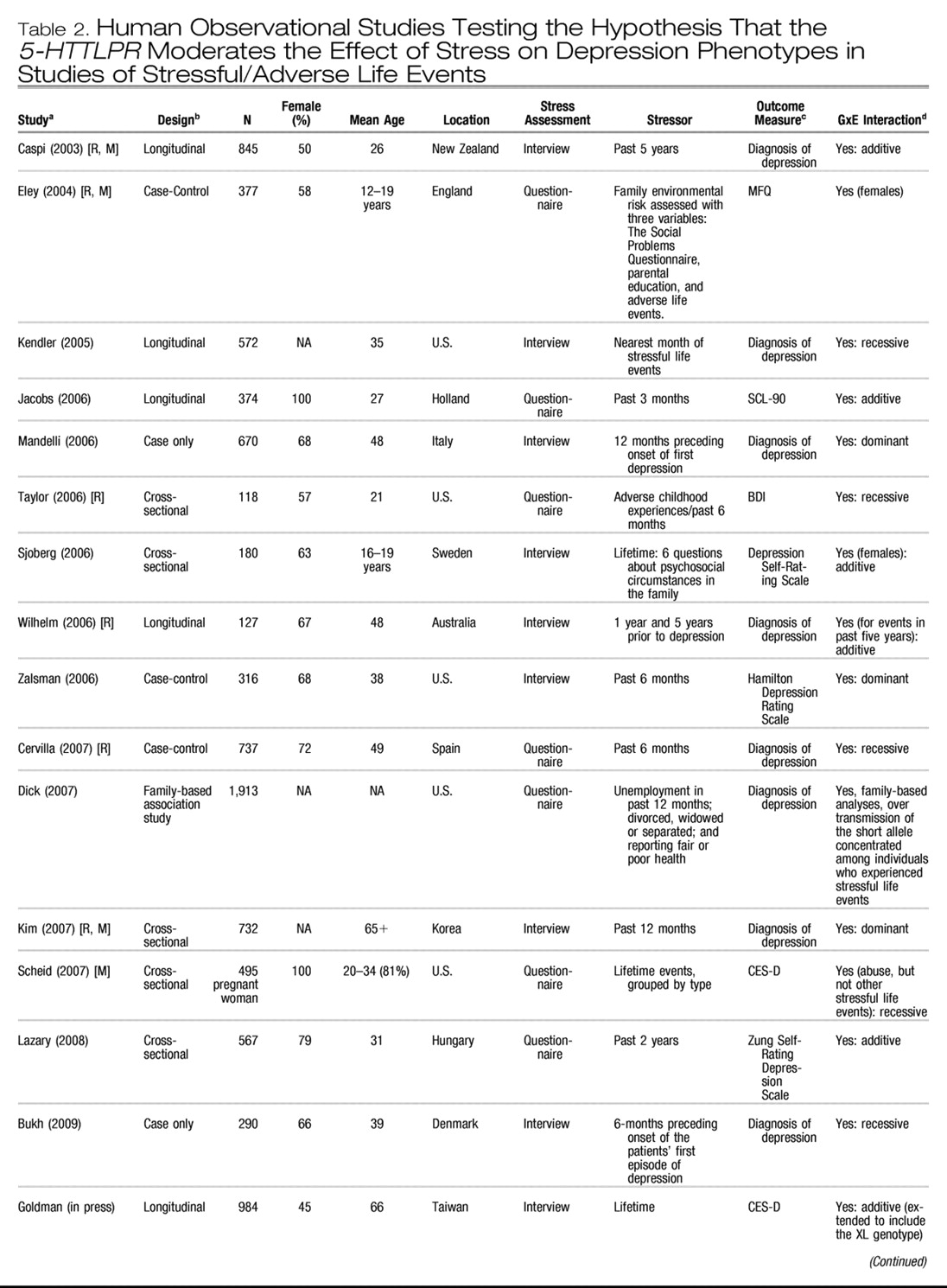
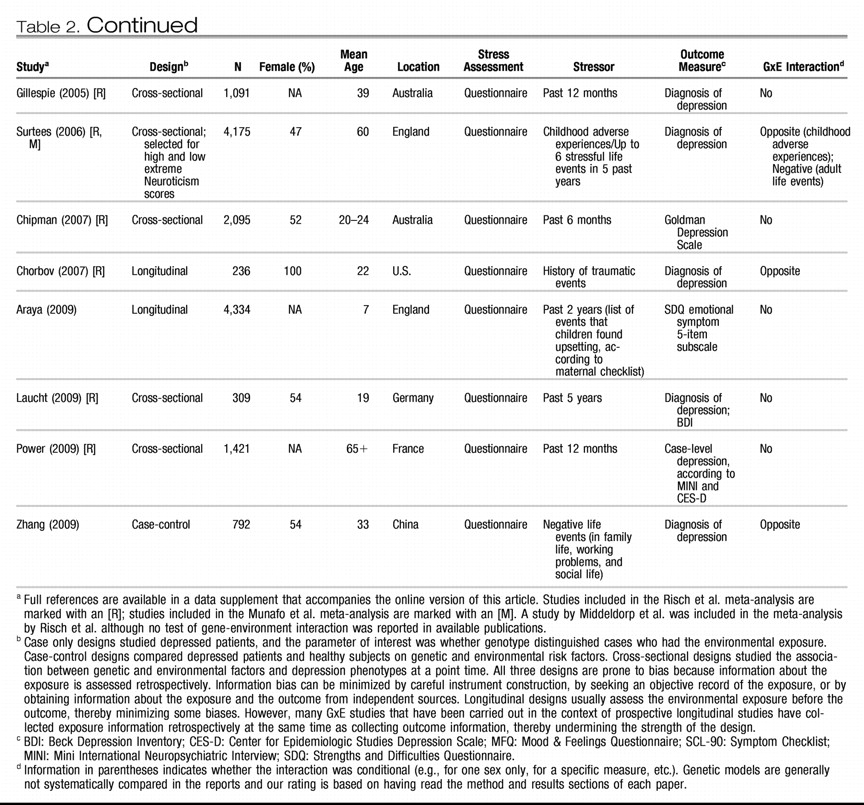
Table 1 and
Table 2 list all human observational studies up to summer 2009 that tested the hypothesis that the
5-HTTLPR moderates the effect of stress on depression phenotypes. Three observations emerge from the tables. First, multiple studies have reported that S-carriage moderates the influence of stress on depression. Whether or not the initial finding can be replicated has been answered in the affirmative. Second, positive findings have emerged from a variety of observational research designs used to test the hypothesis, including phenotype case-only designs, case-control designs, cross-sectional designs, longitudinal designs, and exposure designs. This suggests the finding is “sturdy,” in the sense that its signal can be detected despite noise from varying research settings, sample characteristics, and study designs (
6). Third, there have also been quite a few negative findings. The degree to which negative findings call the original result into question depends on whether differences in study designs are systematically related to differences in study findings. If failures to replicate are characterized by systematically different subject populations or systematically weaker methodologies, their challenge to the original result is greatly diminished.
We considered factors that might covary with positive versus negative findings, including subjects' sex, age, and nationality, and features of phenotype measurement, but these did not covary systematically with findings. However, positive and negative findings did closely track variation in methodological features related to the quality of environmental exposure measurement. Concerns have been expressed about standards of stress assessment in tests of this hypothesis (
7,
8). We call attention to three issues.
First, almost all nonreplications rely on brief self-report measures of stress, whereas studies using objective indicators or face-to-face interviews to assess stress exposure yield positive replications(“Stress Assessment” column in
Tables 1 and
2). Face-to-face interviewers can clarify the meaning of a reported life event and enhance memory for life events by probing and by using techniques such as life event calendars, as did the initial study (
3). In contrast, it is known that self-report event checklists gather idiosyncratic and inaccurate information (
9,
10).
Second, studies of specific stressors consistently yield positive findings. Why are these studies so consistent? One possibility is that their focus on a specific, homogeneous, developmentally relevant, and clearly operationalized depression-inducing event decreased between-subject heterogeneity in the exposure and enhanced internal validity of the study design.
Table 1 groups studies of two specific stressors that are established causes of depression: childhood maltreatment and medical illness. Nine studies report about depression that follows childhood experiences associated with maltreatment and victimization. Although exposure measurement is not uniform, the studies are united by focusing on threatening events in which physical, sexual, or relational harm were carried out or intended. Virtually all of these studies focus on children, adolescents, and young adults. All of them show that S-carriage moderates the association between child maltreatment and depression. Another nine studies report about depression following medical illness. Virtually all focus on middle-aged and elderly participants. Studies of patients suffering hip fractures, strokes, Parkinson's disease, heart disease, and chronic-disease load show that S-carriage moderates the association between medical illness and depression.
Third, whereas studies of specific stressors consistently generate positive findings, studies of stressful/adverse life events yield mixed results (
Table 2). This inconsistency could result from the highly variable measurement of stressful life events (
7,
11). The pool of studies exemplifies five difficulties in stress measurement: 1) Stress measures are sometimes noncomparable and fall prey to the fallacy that because measures have the same name they measure the same construct (
12). For example, some studies count death of a spouse as a stressor, whereas others count being the child of a father in an unskilled job as a stressor. Some studies count stress events, others model event severity. Some stressors are chronic, others acute. Some studies define a “stressor” by its level of distress, others do not. Some studies examine events that happened to the proband, others examine events among the proband's friends and relatives. 2) Some studies assess stress through currently depressed individuals' self-reports, which are biased by mood-congruent memory revision and thus overcount events (
13). Moreover, humans seek explanations, a phenomenon termed “effort after meaning,” which leads respondents who have been depressed to misattribute their illness to a life event. Some studies assess events through long-term retrospective reports (sometimes over decades), which are flawed by forgetting and undercount life events, particularly among respondents who lack depression. In addition, respondents often overcount trivial and undercount severe events (
9). These cognitive processes (mood-congruent memory revision, effort after meaning, and retrospective forgetting) working together can artifactually influence a study's association between life events and depression. Thus, a correlation between life events and depression does not indicate validity, contrary to claims (
14). 3) Some studies test the connection between stress and depression contemporaneously, others across years or decades. 4) Some studies are unable to rule out reverse causation, in which depression precipitates stressful events; for example, one study measured depression over the respondents' lifetime, but ascertained life stress during only the past year (
15). 5) Most studies do not consider variation in participants' depression history, despite evidence that stress is more relevant for initial than recurrent depression episodes.
In the first decade of research about the
5-HTTLPR GxE interaction, scientists have frequently taken advantage of existing data sets, quickly adding genotype data to studies that had previously measured depression and life events for other purposes. Not all of these studies' designs and measures are well-suited to testing the GxE hypothesis. Covariation between poor measurement quality and negative findings was observed early on (
16) and has been confirmed with the increasing number of published GxE studies (
17). Notably, many of the largest studies in
Table 1 and
Table 2 were obliged to collect brief retrospective self-reports of stress through telephone interviews or postal questionnaires in order to contain data collection costs. Thus, unfortunately, large sample size tends to coincide with poor measurement quality, and meta-analyses that give larger samples greater weight in estimating an effect across studies further compound this problem. There is hope that a new generation of cohort studies purpose-built for testing GxE interactions will improve replicability, but these must correct the problems of exposure measurement discussed in the previous paragraph, lest they merely repeat the problems on a far larger scale.
Most observational GxE research on
5-HTT in humans has focused on depression. However, additional evidence links the
5-HTTLPR to a broader range of stress-reactive phenotypes, including PTSD (
18), posttrauma suicide attempt (
19), aggressive reactions to a cold-pressor test (
20), stress-linked alcohol consumption (
21,
22) and substance use (
23,
24), stress-related sleep disturbance (
25), and even premature ejaculation (
26). Research on quantitative endophenotypes shows that S-carriers with high levels of childhood maltreatment and adversity exhibit enhanced anxiety sensitivity (
27) and a bias toward perceiving and expecting negative outcomes (
28). Moreover, S-carrying children who are raised by unresponsive or nonsupportive mothers exhibit poor self-regulation of negative affect (
29–
32), which predicts a variety of adult psychiatric disorders (
33). Finally, research that monitors affective experiences on a daily basis shows that S-carriers experience anxious mood on days with more intense stressors (
34) and larger increases in negative affect while trying to quit smoking (
35). To claim that these diverse outcomes are heterotypic manifestations of a unifying genetic vulnerability to stress reflected in the
5-HTTLPR S allele requires a theory that specifies the unifying mechanism. The leading theory (
1,
36) is that the
5-HTTLPR is a genetic substrate for a latent personality trait, termed negative affectivity or neuroticism. Negative affectivity prospectively predicts risk for all stress-related psychiatric disorders (
37). In theory,
5-HTTLPR S-carriers are characterized by the stable trait of negative affectivity that is converted to psychopathology only under conditions of stress, just as glass is always characterized by the trait of brittleness but shatters only when a stone is thrown. Negative affectivity represents the potential for excitability of anxiety and fear neural circuits, and is characterized by an attentional bias toward negatively valenced information and a cognitive sensitivity to perceive threat (
38). This trait is operationalized in all experimental tests of the
5-HTTLPR GxE hypothesis, reviewed next.
Experimental neuroscience studies
In 2002, a synergy emerged between research in human affective neuroscience and genetic research into the
5-HTTLPR. Specifically, noninvasive functional MRI (fMRI), which assays information processing within distinct neuronal circuits, revealed relatively exaggerated threat-related amygdala reactivity in carriers of the
5-HTTLPR S allele (
2). This initial finding has since been repli-cated in independent samples of both healthy volunteers and psychiatric patients, using a multitude of threatening stimuli and neuroimaging modalities (
39–
49). This effect on the magnitude of amygdala reactivity has recently been extended, with S-carriers also exhibiting a relatively faster response than L-allele homozygotes (
50). Consistent with the heightened sensitivity to environmental threat documented in S-carriers, recent work suggests that the effects of the S allele on amygdala function may be unique to stimulus-provoked amygdala reactivity and not elevated baseline levels of activation (
51–
53).
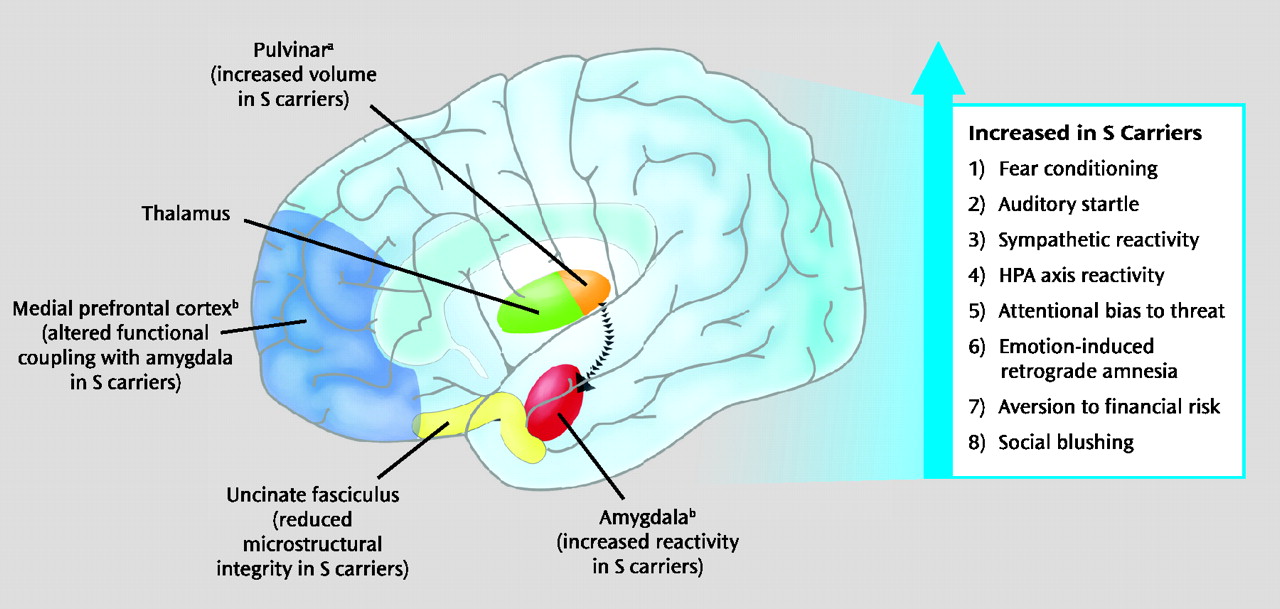
The bias in threat-related amygdala reactivity associated with the
5-HTTLPR S allele is positioned to drive the polymorphism's associations with altered mood and affective disorders, especially in interaction with exposure to environmental stressors and trauma. Evidence from animal and human studies demonstrates that the amygdala mediates both physiological (e.g., autonomic reactivity) and behavioral (e.g., reallocation of attentional resources) effects that allow an individual to respond to environmental and social challenges (
54). Neuroimaging studies have reported positive correlations between indices of anxiety and amygdala reactivity to affective stimuli (especially threatening stimuli) (
55). Such findings demonstrate that variability in the magnitude of threat-related amygdala reactivity predicts individual differences in sensitivity to environmental threat and stress.
Human neuroimaging research suggests that relatively increased amygdala reactivity associated with the
5-HTTLPR S allele is likely to reflect both the functional and structural architecture of a distributed network of brain regions. Research suggests that this network communicates information about the environment to the amygdala and relays signals between the amygdala and regulatory circuits in the medial prefrontal cortex. This putative mechanism is further underscored by the significant role serotonin signaling plays in the general development and function of this extended neural network (
56). The S allele has been associated with altered functional coupling (as indexed by correlated fMRI signal strength) between the amygdala and regions of the medial prefrontal cortex (
40,
57). These medial prefrontal regions integrate amygdala-mediated arousal and down regulate amygdala reactivity. Medial prefrontal regions are also involved in the extinction of conditioned fear responses, which are dependent on amygdala circuitry.
The pattern of
5-HTTLPR-associated differences in the functional dynamics of the amygdala and medial prefrontal cortex is echoed in structural measures within this same network. Specifically, the S allele has been associated with relatively decreased gray matter volume in the amygdala and medial prefrontal cortex (
42,
57). The S allele has also been associated with alterations in the microstructure of the uncinate fasciculus, the white matter fiber bundle providing the majority of connections between the amygdala and medial prefrontal cortex (
58). Individual differences in uncinate fasciculus microstructure correlate with trait anxiety (
59). In addition, postmortem tissue analyses have associated the
5-HTTLPR S allele with relative enlargement of the pulvinar, which relays visual information to subcortical and higher cortical brain regions (
60). Consistent with this, as well as with amygdala-mediated behavioral arousal, numerous studies have reported increased cortical activity in response to experimental provocation in S-carriers (
61–
66).
In addition, a growing group of studies has begun to document effects of the
5-HTTLPR on intermediate behavioral and physiological processes that map onto these alterations in brain structure and function. The S allele is associated with increased acquisition of conditioned fear responses (
67), increased auditory startle response (
68,
69), and greater sympathetic reactivity when simply observing another person receiving shock (
70). Moreover, the
5-HTTLPR S allele has been associated with increased HPA axis reactivity to aversive or threatening stimuli in a number of studies (
71–
74). The S allele typically has no impact on baseline levels of HPA function in these studies, underscoring its documented effect on threat-related amygdala reactivity. In addition, the S allele has been linked with difficulty disengaging from, or preferential attention toward, threat-related stimuli (
75–
79), a more negative information-processing bias (
80), emotion-induced retrograde amnesia (
81), sensitivity to financial loss (
70,
82), and even social blushing (
83). Although this literature is not without inconsistencies (e.g., some reported associations are sex-specific and others have not replicated), it does suggest that the effects of the
5-HTTLPR S allele on the brain's neural circuitry for responding to environmental threat and stress translate to biases in both behavioral and physiological processes which may, in turn, shape individual risk for depression upon exposure to acute trauma or chronic stressors (
Figure 2). Multiple components of this ongoing research were highlighted in one report of increases in threat-related amygdala and medial prefrontal cortical activation as well as heart rate and startle amplitude in
5-HTTLPR S-carriers who also exhibited a self-reported sensitivity to perceived danger in the environment (
28).
Nonhuman primate studies
Rhesus monkeys have an orthologue of the human
5-HTTLPR, making them an excellent model species for GxE studies. Like the human variant, the rhesus S allele is associated with decreased transcriptional efficiency in vitro (
84). The modulating influence of the polymorphism on early life stress has been tested by separating infant rhesus monkeys from their mothers and rearing them with other infants (a long-established model of early life adversity in this species). During initial episodes of separation, monkeys carrying the
rh5-HTTLPR S allele exhibit less “protest” and self-directed behaviors that are considered active coping responses to this stressor (
85). Instead, separated S-allele monkeys display greater anxiety, agitation, stereotypies, and an exaggerated HPA axis response (
85,
86).
The modulating influence of the
rh5-HTTLPR on separation in infancy persists into later life, manifesting, for example, as higher ACTH responses to stress in S-carrier monkeys than LL homozygotes (
87). It is important to underscore that these long-lasting phenotypic effects of the S allele only occur in monkeys exposed to maternal separation early life stress, echoing the GxE interaction observed in relation to human depression.
Another major parallel between the human and monkey data has been the finding that, as in humans, the stress-related S allele phenotype in monkeys is related to an intermediate neural phenotype characterized by abnormal corticolimbic structure and function. For example, the S allele in monkeys also has been mapped onto reduced gray matter volumes in the amygdala, medial prefrontal and orbitofrontal cortex, and pulvinar (
88). Moreover, monkeys with the S allele exhibit greater metabolic activity than LL homozygotes in the amygdala and its networked cortical regions, including orbitofrontal cortex, in response to the stress of relocation (
89). Given the importance of the orbitofrontal cortex in social behavior, abnormalities in this region might also account for the finding that S-carriers engage in less eye gaze with high status conspecifics and are more risk-averse in their presence (
90). An intriguing development is recent data from S-carrier monkeys (
88) and
5-HTT mutant mice (
91) demonstrating that reversal learning, a measure of cognitive flexibility subserved by the orbitofrontal cortex (
92), is enhanced as a function of relative
5-HTT gene deficiency. This may reflect increased sensitivity to negative environmental stimuli, although further work will be needed to substantiate this. Notwithstanding, these data indicate that altered
5-HTT gene function may influence multiple higher behaviors, as would be predicted if it affects a core corticolimbic neural circuitry.
Studies involving genetically engineered 5-HTT mutations in rodents
Research using rodents allows for experimental control over genetic background and the environment to a degree that is neither practically nor ethically feasible in human or even nonhuman primate studies (
93). Although there is functional gene variation in the murine
5-HTT (slc6a4) (
94), there is no rodent orthologue of the
5-HTTLPR. As an alternative approach, mice and rats have been genetically engineered with loss-of-function mutations in the
5-HTT gene. Studying the consequences of these mutations for behavior and brain function has greatly complemented the work on the
5-HTTLPR in primates and provided some key insights into the mechanisms that mediate the influence of
5-HTT on negative affect and stress reactivity (
56,
95).
Mice in which the
5-HTT has been functionally excised either by targeted mutation or chemical mutagenesis exhibit heightened anxiety-like behavior, impaired fear extinction, and exaggerated HPA-axis responses to acute stress. While it is far less common to engineer mutant rats than mice, a
5-HTT-null mutant rat has been generated and also shows increased anxiety-like behavior (
96). Furthermore, providing an interesting counterpoint to these “knockout” mutants, mice with transgenic overexpression of the
5-HTT actually produce decreased anxiety-like behavior (
97). The consistency of these findings across models, laboratories, and species is rarely seen in the field of rodent behavioral genetics and illustrates the strong penetrance of the mutation's effects.
The “depression-related” consequences of rodent
5-HTT knockout mice are, at first blush, less consistent than the anxiety-related consequences, in that they are seen in some of the standard rodent assays for this behavior but not others. This variability may, however, be a legitimate reflection of differences in the level of stress evoked under varying test conditions. In support of this hypothesis, following repeated exposure to stress (e.g., forced swimming, tail suspension),
5-HTT knockout mice develop a depression-related “despair-like” phenotype that is not seen with single exposure (
97). The parallels with the primate data showing that the S-allele influence on depression is contingent upon repeated stress exposure are clear.
Much of our understanding of the functional role of the
5-HTT as a master modulator of the 5-HT system has been built upon work in rodents (
98). As such, researchers have a ready platform and toolset from which to perform certain neural and molecular analyses in
5-HTT mutant rodents (e.g., in vivo measurement of brain 5-HT availability) that cannot be employed in humans. One of the key themes to emerge from this work is that the neural consequences of
5-HTT gene mutation extend well beyond the
5-HTT and its role as a regulator of 5-HT availability.
5-HTT null mutation leads to alterations throughout the 5-HT system that include changes in 5-HT receptor binding and 5-HT synthesis (
56,
95). At the systems level,
5-HTT knockout mice exhibit an abnormally high density of excitatory dendritic spines on amygdala neurons and an increase in dendritic arborization of prefrontal cortex neurons (
56). The implication here is that influence of
5-HTT variation may not be limited to effects on 5-HT availability or even on the 5-HT system. Recently, this implication was confirmed in a rhesus macaque model (
88), in which the
rh5-HTTLPR S allele affected behavior and brain morphology but not
5-HTT (
99) or 5-HT
1A concentrations in vivo. Similar complexities in the likely molecular consequences of the
5-HTTLPR have been documented in humans (
100–
105). Collectively, mouse and monkey and human findings suggest that
5-HTTLPR's behavioral effects on stress-reactivity may be most consistently rooted in neural development.
An intriguing line of enquiry in this context has centered on the hypothesis that
5-HTT variation may in part modulate the capacity to cope with stress by shaping the early life development of corticolimbic circuitry (
56). In fact, the importance of the 5-HT system in neurodevelopment has long been recognized, and the
5-HTT is known to be critical for the formation of cortical systems in particular (
106,
107). Pharmacological inhibition of the
5-HTT during early life mimics the anxiety-like phenotype of
5-HTT knockout (
108). Moreover, poor maternal care produces heightened anxiety-like behavior in mice with a partial (heterozygous)
5-HTT null mutation, which are phenotypically normal under conditions of good maternal care (
109).
These findings raise the question of whether the effects of
5-HTT knockout are developmentally driven. It has been hypothesized that the
5-HTTLPR GxE interaction observed in relation to adult stressful life events should selectively affect people already “primed” by childhood adversity (
8). This opens up some very interesting avenues for future animal studies. For example, would
5-HTT loss restricted to early life development be sufficient to increase anxiety and impair stress-coping? If so, is there a critical window and what is the corresponding ontogenic period in humans? Researchers could then elucidate the key neural and molecular changes underlying these effects. This could, in turn, “square the circle” by nominating mechanisms to target with novel therapeutic approaches in humans.