INTRODUCTION
Most theories of drug-addiction mechanisms have been based on animal models and, until recently, these theories have made the assumption that all subjects are alike in their responses to drugs (
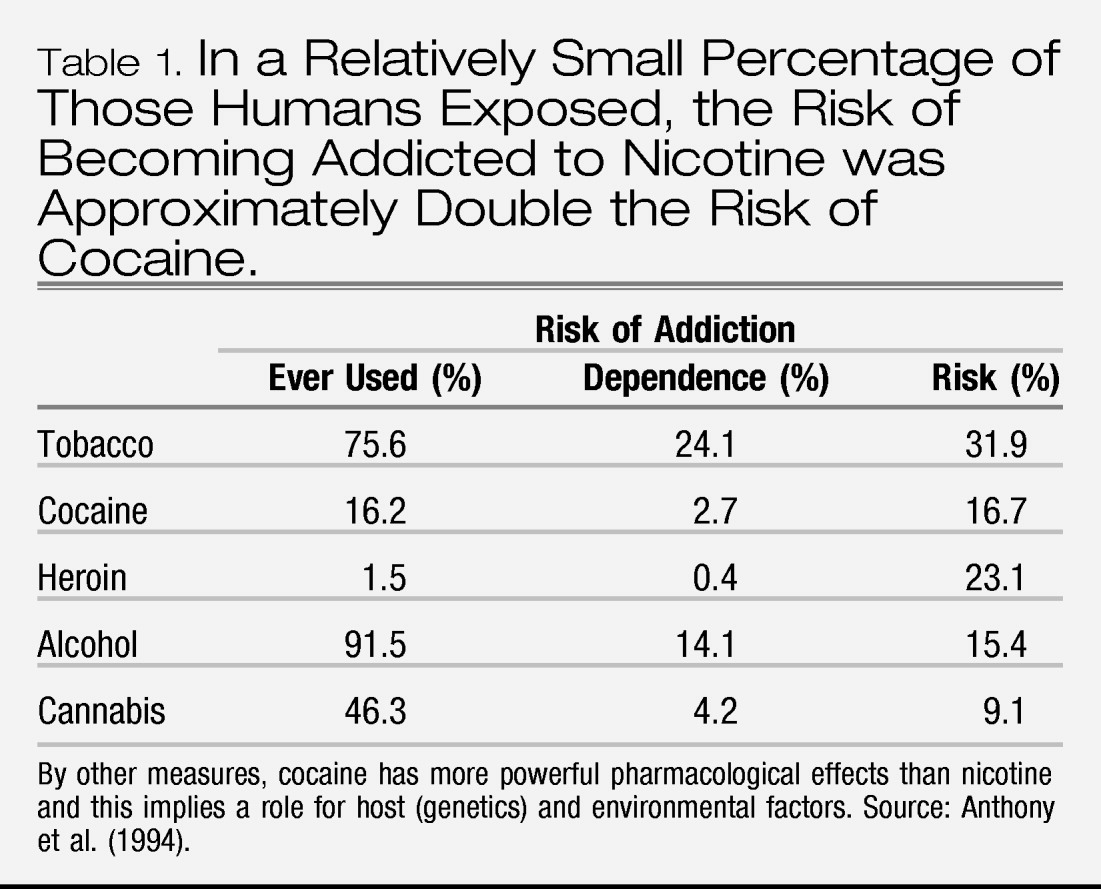
Deroche-Gamonet et al. 2004). In reality, human subjects are quite variable in how they respond to drugs. Moreover, data from the studies of non-human primates indicate that genetic variation is also important in other higher species. Drugs that demonstrate rewarding properties in animals also tend to be abused by humans, but only by a relatively small percentage of those humans exposed (
Table 1). The most obvious effects of chronic drug use are tolerance and physiological dependence and these phenomena translate well from animals to humans. However, tolerance and its complement, physiological dependence, are normal reactions and do not imply addiction.
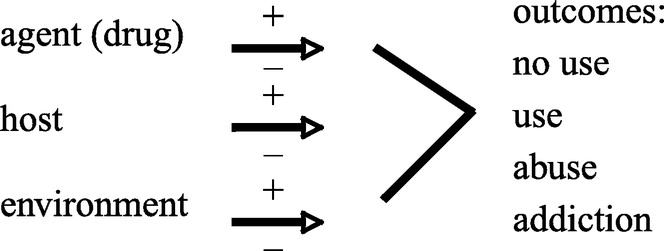
At the clinical level, the theoretical model of addiction is similar to that of an infectious disease such as tuberculosis. The development of addiction depends on the interaction of agent, host and environment (Fgure 1). The progression from use to abuse to addiction is determined by this interaction. An understanding of addiction requires addressing all three of these classes of variables. Treatment and prevention efforts that fail to consider all three have not been successful. Therefore, it follows that pharmacological treatments must be imbedded in a comprehensive rehabilitation programme that addresses these variables to the extent possible. While first use is under cognitive control, albeit usually influenced by social pressures, the user with genetic vulnerability will progress to a stage of compulsive use. At this point, volitional control is greatly diminished. As an example, the reader need only think of friends and relatives who repeatedly relapse to compulsive cigarette smoking despite the knowledge of the hazards to their health and despite having expressed a conscious desire to abstain from smoking. At the neuronal level, this compulsive relapse suggests plasticity, a memory trace. No treatment has even been theorized, however, that could selectively ‘erase' addiction memories while not impairing adaptive memories and new memory formation.
The mechanisms involved in the production of euphoria vary according to the pharmacological category of the drug and have been discussed in earlier papers of this issue. All have in common the activation of brain reward pathways that have evolved to ensure survival, which largely explains the compelling nature of drug reward. The activation of the reward system produces reinforcement of drug acquisition behaviours and associations between environmental cues that signal the arrival of drug effects or drug withdrawal symptoms as drugs disappear from the body through metabolism. The environmental stimuli that become neurologically associated with drug effects are variable and may include persons, places and situations. Treatment approaches, therefore, have attempted to diminish the strength of these conditioned reflexes that lead to relapse and facilitate the development of new memories that produce natural rewards. In this paper, pharmacological approaches that reduce aversive effects of drug cessation, reduce drug reward or reduce drug desire or craving will be discussed. Behavioural approaches are also necessary in combination with medication, but will not be discussed here.
The modern definition of addiction emphasizes uncontrolled drug use rather than tolerance and physiological dependence as essential features of the disorder. It is generally recognized that addiction has strong hereditary influences and once established, it behaves as a chronic brain disorder with relapses and remissions over the long term (
McLellan et al. 2000). The diagnostic criteria, which are signs of compulsive drug seeking, are the same for all drug categories (ethanol, opioids, stimulants, sedatives, nicotine and cannabinoids), even though the mechanisms for activating the reward system are quite different.
With active drug use, euphoria alternates with craving to establish a cycle of addiction that becomes increasingly entrenched and uncontrollable, despite medical and psychosocial hazards. Craving (a strong desire for a drug) can be precipitated by environmental cues, stress or exposure to a small dose of the addictive agent (priming). Cue-induced craving has been associated with limbic system activation in a large number of human neuroimaging studies using positron emission tomography and functional magnetic resonance imaging (
Childress et al. 1999), and this pernicious and persistent phenomenon might be reversed by agents that dampen limbic activation. Human neuroimaging studies also demonstrate reductions in frontal lobe metabolism with stimulant, opioid and alcohol dependence, thus providing a possible explanation for deficiencies in behavioural inhibition among addicts (
Franklin et al. 2002). Numerous animal studies indicate that chronic exposure to addictive agents disturbs reward function (
Koob et al. 2004). These findings support a biological basis for addiction and are guiding neuronal strategies that target specific clinical components of addiction.
The clinical data fit best when addiction is considered to be a syndrome characterized by compulsive drug-seeking behaviour that impairs psychosocial functioning or health. Even after detoxification and long periods of abstinence, relapse frequently occurs despite sincere efforts to avoid further drug use. People or situations previously associated with drug use elicit involuntary reactions and may provoke relapse (
Wikler 1973;
O'Brien et al. 1975). The biological mechanisms for these reflex patterns are suggested by the data from animal models at the neurochemical and molecular levels, as discussed in this issue. At the clinical level, conditioned cues produce intense craving through involuntary limbic activation, leading to self-destructive drug use even after long periods of abstinence. In cocaine addicts, even a brief exposure (33 ms) to drug cues evokes limbic system activation (
Childress et al. 2008). A key point for the clinician to realize is that the proneness to relapse is based on changes in brain function, which continue for months or years after the last use of the drug. Of course, these changes in brain function interact with environmental factors such as social stress and situational triggers.
If tolerance and withdrawal symptoms were the only elements of addictive illness, treatment would simply consist of detoxification, a process that allows the body to cleanse itself while receiving descending doses of a medication that reduces withdrawal symptoms (
O'Brien 2006). If drug taking does not resume, homeostatic mechanisms will gradually readapt to the absence of the drug (
LeBlanc et al. 1969) and tolerance will be diminished or lost. We now know that detoxification is, at best, a first step in treatment and that simply achieving a drug-free state is not the most significant accomplishment. The more difficult aspect is the prevention of relapse to drug-taking behaviour.
DETOXIFICATION
It is unfortunate that the majority of drug-dependent persons are merely treated with detoxification and little or no long-term follow-up care. This is not logical, but it is a fact of the current health-care system in the USA (
McLellan et al. 2005). Detoxification is actually performed by the patient's own metabolic processes. Thus, it can be accomplished voluntarily (although not necessarily safely) through sheer will power by ceasing drug use or accomplished involuntarily when an addict is incarcerated or placed in a treatment programme where access to drugs is denied. The withdrawal syndrome from opiate addiction can be very uncomfortable, but it is not life-threatening unless the patient has pre-existing medical problems. The symptoms consist of sweating, muscle aches, cramps, nausea, diarrhoea, vomiting, lachrymation, rhinorrhoea, tremors, tachycardia and other signs of autonomic nervous system hyperactivity. The discomfort has been compared to a bad case of the flu. Several sorts of treatment of these symptoms are available. Withdrawal from sedatives, alcohol and stimulants will be considered below.
Replacing the drug of dependence or using another drug in the same pharmacological category in gradually decreasing doses is a direct way to block withdrawal symptoms. As in all forms of detoxification, transfer from a short-acting drug such as heroin to a longer acting drug such as methadone provides a smooth transition to the drug-free state. By appropriate dosing, detoxification can be achieved with minimal discomfort. A recent innovation for opiate dependence involves using the partial agonist buprenorphine as a transition to the drug-free state. The patient can be switched from dependence on heroin or methadone to buprenorphine, which is then stopped with few or no withdrawal symptoms.
The same principles apply in the detoxification from nicotine dependence using nicotine replacement and from sedative (ethanol) dependence using another sedative such as a benzodiazepine. Stimulant (cocaine and amphetamine) withdrawal does not usually require medication, but rapid return to drug use is frequent. A medication that reduces stimulant withdrawal symptoms such as modafinil (see paragraph, Blockade of euphoria) may reduce relapse.
In the treatment of patients dependent on alcohol or other sedatives, appropriate detoxification is critical because the sedative withdrawal syndrome is potentially life-threatening. Whereas the acute administration of alcohol and sedatives increases γ-aminobutyric acid (GABA) and decreases glutamate activation, the reverse occurs with chronic exposure, producing a GABA deficiency state and glutamate hyperactivity that increases the risk of seizures during withdrawal (
Dackis & O'Brien 2003). There is evidence that sensitization to alcohol withdrawal symptoms occurs, so repeated withdrawals become progressively more severe. The treatment of withdrawal symptoms with benzodiazepines may retard the sensitization process (
Brown et al. 1988). Benzodiazepines effectively suppress the sedative withdrawal syndrome, and with proper attention to electrolytes and vitamins, the vast majority of patients can be safely eased into the alcohol-abstinent state in preparation for a long-term rehabilitation programme.
Symptoms of nicotine withdrawal can be diminished by nicotine replacement therapy through chewing gum, patch or nasal spray. Nicotine gum and nicotine patch do not achieve the peak plasma levels seen with cigarettes, and thus they do not produce the same magnitude of nicotine's pleasant effects. Comparisons with placebo treatment show large benefits for nicotine replacement at six weeks, but the advantage diminishes with time.
The withdrawal syndrome from stimulants such as cocaine and amphetamine consists of hypersomnia, hyperphagia, bradycardia and a number of depressive symptoms that usually resolve over several days. Interestingly, cocaine withdrawal symptoms appear to be predictive of treatment outcome.
Kampman et al. (2001) measured cocaine withdrawal symptoms in several trials using the Cocaine Selective Severity Assessment. They found that more withdrawal symptoms in subjects at the start of treatment accurately predicted poorer outcome following treatment. Given these findings, the pharmacological reversal of cocaine withdrawal symptoms with agents such as modafinil may improve clinical outcome (
Dackis et al. 2005).
Heavy marijuana users also develop a physical dependence and may present for treatment when they are unable to stop daily use on their own (
Haney et al. 2004). The symptoms consist of irritability, anxiety, marijuana craving, decreased quality and quantity of sleep and decreased food intake. Various medications have been used to alleviate these symptoms and some clinicians have reported success with dronabinol, the oral form of delta-9-tetrahydrocannabinol, but controlled clinical trials are lacking.
Detoxification by the suppression of autonomic hyperactivity
For opiate detoxification, methadone is not always available due to legal limitations and buprenorphine may be undesirable because it is a partial opiate agonist. Clonidine, an α
2-agonist, reverses opiate withdrawal by acting on autoreceptors producing presynaptic inhibition of locus coeruleus activity. This effectively reduces the large adrenergic component of opioid withdrawal (
Gerra et al. 2001). Lofexidine, a similar medication, has been successfully used in the UK as an aid to opiate detoxification and is in clinical trials in the USA. Thus, clonidine and lofexidine have found a place in the clinic for treating the symptoms of opioid withdrawal. Very rapid detoxification under general anaesthesia has also been used, but there are no data to support an advantage over standard treatment.
PRINCIPLES OF RELAPSE PREVENTION
As a chronic disorder, addiction requires long-term treatment that should be measured in months and years. The strategies for preventing relapse have traditionally involved counselling or psychotherapy and, more recently, include pharmacotherapies that target clinical components of addictive illness. When psychiatric disorders co-occur with addiction, these disorders must be treated concomitantly and preferably by the same treatment team. This paper focuses on the medication for the primary addictive disorder, but counselling and medication of co-occurring disorders are equally important.
The treatment of addicted patients must always be individualized. This requires a complete evaluation so that coexisting medical, psychiatric and social problems can be addressed as needed. There are, however, common elements to treatment programmes. Treatments for addictive disorders may begin with detoxification, but the key to successful treatment is the long-term prevention of relapse by behavioural and pharmacological means. Usually, these approaches should be combined; insistence on behavioural treatment alone and without medication remains one of the chief weaknesses in many treatment programmes. The types of medication that have shown efficacy in combination with behavioural therapy in the prevention of relapse can be classified as agonists (including partial agonists), antagonists and anti-craving medications that work through a variety of mechanisms. Vaccines are an experimental approach that is currently being evaluated in clinical trials (
Martell et al. 2005;
Sofuoglu & Kosten 2006).
Agonists and partial agonists
The first use of an agonist for the treatment of addiction was reported in the 1960s by Vincent Dole and colleagues who demonstrated that daily methadone could transform the behaviour of opiate addicts, reducing craving and permitting the addict to engage in productive activities (
Dole & Nyswander 1965). Opiates (which are derivates of the opium poppy) and opioids (which may be peptides or synthetic compounds) activate opiate receptors that are located throughout the nervous system, as well as in the endocrine, cardiovascular, gastrointestinal and other systems of the body. The behavioural effects of opiates include intense euphoria and calming. The user becomes satisfied and relaxed, a state quite different from the euphoric excitement produced by stimulants. In the presence of pain, opiates and opioids produce analgesia, so there is both relaxation and relief of severe pain.
While opioids produce prompt physiological dependence with repeated use, addiction seldom occurs in patients receiving opioids for the relief of severe pain (
Adams et al. 2006). Of course, prescription opiates and opioids can be abused and it is the responsibility of the physician to provide humane pain relief to patients in need while exercising caution to reduce the likelihood that the prescribed medication will be obtained by deliberate abusers. The use of heroin or other opiates purchased on the street for the purpose of obtaining a ‘high' has a significant risk of producing addiction.
Detoxification is not applicable to those opioid-dependent patients who prefer transition to maintenance using methadone or buprenorphine. Methadone has a slow onset by the oral route. It is a long-acting μ-opiate receptor agonist that largely prevents reward or euphoria if the patient ‘slips' and takes a dose of an opiate. The mechanism for preventing euphoria is based on cross-tolerance in which tolerance (insensitivity) acquired by the use of one drug in a category conveys tolerance to all drugs in that category. Of course, the maintenance dose of methadone must be adjusted to the purity of heroin on the street. A dose of heroin significantly higher in opioid equivalents than the maintenance dose of methadone would override the cross-tolerance effect. Patients can be maintained for many years on a properly adjusted dose of methadone. Craving for opioids is diminished or absent, and patients are able to engage in constructive activities. Cognition and alertness are not impaired, and complex tasks including higher education can be accomplished (
Kreek 1992). Currently, approximately 200,000 former opiate addicts are being maintained on methadone in the USA. Those with significant psychosocial problems require counselling or psychotherapy in addition to the medication.
As a partial agonist, buprenorphine produces limited opiate effects, and thus overdose is rare except when combined with benzodiazepines. Owing to its high affinity for the μ-receptor, buprenorphine effectively prevents access to the receptor by other opiates and opioids, thus reducing the likelihood that other opioids will be used. Patients treated with buprenorphine become physiologically dependent on it, as is the case with methadone, but if buprenorphine is stopped, withdrawal symptoms are quite mild. A limitation of buprenorphine is the ceiling on opiate agonist effects giving a maximal efficacy equivalent to approximately 40–50 mg of methadone. Addicts using large doses of street heroin may find that buprenorphine is not sufficiently potent to block withdrawal or drug craving.
Nicotine replacement has been listed under “
Detoxification” in this article, as a treatment of nicotine withdrawal. The administration of nicotine as a patch, gum or nasal spray can also be used as a maintenance treatment for extended periods as is the case with methadone. The levels obtained via a nicotine patch usually do not produce the pleasant responses achieved through smoking and there are usually no withdrawal symptoms on stopping the patch. Theoretically, smokers should be able to switch their nicotine dependence from administration via smoking to nicotine delivered by patch, chewing gum or nasal spray. Although some smokers continue to chew nicotine gum for many months after giving up cigarettes, most discontinue nicotine replacement after a few weeks. The tendency to relapse may be strong, and thus it is important to teach patients behavioural techniques to resist the urge to smoke. An interesting approach that showed some efficacy in clinical trials is the combination of nicotine and mecamylamine, a nicotinic receptor antagonist, to prevent relapse to smoking. It was hypothesized that stimulation of receptors by both an agonist and an antagonist would be more effective, and the side effects of the two drugs would tend to cancel each other (
Rose et al. 2001). The clinical data on this combination have been mixed, and more studies are needed.
Clinical serendipity played a role in the discovery of bupropion as an effective treatment of nicotine dependence (
Ferry 1994). Originally used as an antidepressant, bupropion was found to significantly improve abstinence rates in smokers whether or not they were depressed (
Swan et al. 2003). The mechanism is unclear, but one effect of bupropion is the relief of negative affect in recently abstinent smokers (
Lerman et al. 2002).
A very recent medication that applies the partial agonist principle in the treatment of tobacco use disorder is varenicline (
Foulds 2006). This is an α4 β2 nicotinic receptor partial agonist that has been reported to relieve cigarette craving and to result in a significantly higher rate of abstinence at 52 weeks (23%) than placebo (10.3%) or bupropion (14.6%) in company-sponsored double-blind trials (
Jorenby et al. 2006).
Although agonist treatment is effective in opioid and nicotine addiction, agonists have not been found effective in patients addicted to stimulants. Experiments using methylphenidate or dextroamphetamine as agonists for cocaine and methamphetamine addiction have not been successful (
Gorelick et al. 2004). Similarly, benzodiazepine treatment for alcohol or sedative dependence is ineffective. It is unclear why agonist treatment does not work in patients addicted to these substances.
Antagonist treatment
Advances in understanding how opioids interact with opiate receptors to produce their pharmacological effects led to the development of specific antagonists that have high affinity for these receptors, but do not activate the chain of cellular events producing opioid drug effects. Naltrexone is an antagonist that has great affinity for μ-opiate receptors and significant but less affinity for ∂- and κ-opiate receptors. Unlike methadone, it has no agonist effects, so there are no opioid calming or other subjective effects. When first introduced, naltrexone was thought to be an ideal medication for heroin addiction because it occupied opiate receptors and blocked the effects of subsequent heroin injections. Experience has shown that most heroin addicts prefer methadone because it provides mild opioid-reinforcing effects that are absent in naltrexone. Thus, naltrexone has been used very little except for white-collar opiate addicts such as physicians, nurses and former addicts released from prison on probation (
Cornish et al. 1997). The effects of blocking opiate receptors probably depend on the degree of tonic activation of the endogenous opioid system. Some normal volunteers given naltrexone experience nausea and dysphoria, while others experience no reaction. Although long-term blockade of opiate receptors might be expected to produce impairment of neuroendocrine function, remarkably few effects have been noted even in patients who have taken naltrexone daily for several years.
In 2006, a slow-release (depot) injectable preparation of naltrexone was given FDA approval and made available for prescription. Paradoxically, it was approved only for alcoholism because it was discovered in animal models that alcohol activated endogenous opioids (
Altshuler et al. 1980). Subsequent work has clearly shown that endogenous opioids are involved in alcohol reinforcement. This was a discovery in an animal model that led directly to a completely novel treatment for alcoholism in humans. Such translational examples give hope that knowledge gained from basic research will eventually lead to new and better treatments. In 1983, clinical trials began and it was found that blocking opiate receptors with naltrexone significantly reduced relapse to clinically significant drinking (
Volpicelli et al. 1990,
1992).
Of course, the depot form of naltrexone is also effective for the treatment of opioid addiction (
Comer et al. 2006), and clinical trials are underway that will eventually lead to FDA approval for that indication in addition to alcoholism. Thus, opiate receptor antagonists have been found to be effective in the treatment of both opiate addiction (blocking external opiates) and alcoholism (blocking endogenous opioids). The availability of a depot form is expected to significantly improve the adherence to medication for this treatment method.
Genomic subcategories
The evidence for genetic influence on vulnerability to addiction is strong, but as with other complex psychiatric disorders, gene association studies based on diagnosis have not identified consistent susceptibility genes. Diagnosis in psychiatry still depends on behaviour rather than biomarkers. Recently, a sub-category of alcoholism based on a functional allele of the gene for the μ-opiate receptor has been studied as a candidate gene for an alcoholism endophenotype. The critical functional observation is an increased stimulation effect from alcohol in carriers of this gene. Recent reports of significantly improved treatment results in alcoholic patients selected on the basis of this genetic variant have raised the exciting prospect of alcoholism treatment guided by genomic testing (
O'Brien 2008).
The variant is an A to G substitution at position 118 of exon 1 of the μ-opioid receptor gene and results in a receptor that has been reported to have greater affinity for β-endorphin (
Bond et al. 1998). Individuals with this allele have been found to perceive greater stimulation from a given dose of alcohol (
Ray & Hutchison 2004) and the stimulation was found to be blocked by naltrexone pretreatment (
Ray & Hutchison 2007). Reduced euphoria from alcohol was also reported in clinical trials of alcoholics randomized to naltrexone and in heavy-drinking volunteers with a family history of alcoholism given alcohol in the laboratory. A retrospective analysis of alcoholic patients in a naltrexone clinical trial found that those with the G allele did poorly when randomized to placebo, but had a significantly better outcome when randomized to naltrexone (
Oslin et al. 2003). This finding was replicated in another clinical trial (
Anton et al. 2008), but not in a small sample of US Veterans Affairs alcoholics (
Gelernter et al. 2007).
Taken together, the various lines of evidence suggest that alcohol activates the endogenous opioid system and this activation is exaggerated in carriers of this genetic variant. The proposed circuitry involves β-endorphin neurons that modulate ventral tegmental GABA neurons inhibiting dopamine (DA) neurons. Alcohol causes a release of β-endorphin that inhibits GABA neurons, thus releasing DA neurons from inhibition and allowing dopaminergic stimulation. This mechanism is supported by microdialysis studies showing that the alcohol-induced DA increase is blocked by naltrexone (
Gonzales & Weiss 1998).
Blockade of euphoria
There are different mechanisms whereby medications can block or diminish the euphoria produced by drugs of abuse. Antagonist treatment prevents the addictive agent from effective binding to brain receptors that mediate the euphoric response. This is best illustrated by naltrexone treatment in opiate dependence, since the mechanism of opiate euphoria results from the stimulation of μ-opiate receptors. The mechanism of stimulant euphoria is not well understood, so it has been more difficult to develop medications to block this essential clinical phenomenon. Nicotine replacement therapy for smokers can reduce the pleasure of smoking by a cross-tolerance mechanism similar to that of methadone for heroin euphoria.
Modafinil, a medication that retards sleep onset, has been shown to block cocaine-induced euphoria in three human laboratory studies of predominantly male subjects (
Dackis et al. 2003;
Malcolm et al. 2006;
Hart et al. 2007). The mechanism of this blockade is unknown. Other medications block cocaine reward in animal studies (but not yet reliably demonstrated in humans) by increasing GABA-inhibitory effects on reward pathways such as ventral tegmental-ventral striatal DA pathways. These include medications such as topiramate (
Kampman et al. 2004), vigabatrin (
Brodie et al. 2005) and baclofen (
Weerts et al. 2007). Animal models suggest that this GABA-enhancing mechanism may block the rewarding effects of alcohol as well as cocaine.
Medications that produce an aversive response
Until 1995, disulfiram was the only medication available to prevent relapse to uncontrolled drinking in detoxified alcoholics. This medication blocks the metabolism of alcohol, causing the accumulation of acetaldehyde, a noxious by-product. The resulting acetaldehyde reaction is so unpleasant that it effectively prevents patients from consuming any alcohol. Disulfiram has a place in the pharmacopoeia of medications for alcoholism but its usefulness is limited. Despite treatment contracts and even legal coercion, most alcoholics will not take disulfiram regularly and randomized clinical trials have not shown disulfiram to be efficacious (
Fuller et al. 1986). Aversive conditioning has also been tried by timing an injection of emetine to produce vomiting while presenting the smell and taste of alcohol (
Childress et al. 1985). A short-term success has been reported, but the technique has never become widespread.
Anti-craving medications
The pharmacological reversal of clinically significant neuroadaptations has long been employed with detoxification regimens, and the normalization of more persistent neuroadaptations might identify agents with anti-craving action (
O'Brien 2005). In addition to chronic neuroadaptations in reward-related pathways, prefrontal cortical dysregulation has been demonstrated in stimulant, opioid and alcohol dependence (
Dackis & O'Brien 2005), and is now viewed as a core component of addiction that contributes to poor impulse control, reduced motivation and denial in addicted individuals. Consequently, agents that enhance prefrontal cortical metabolism hold promise in the treatment of addiction.
Acamprosate, a medication that appears to decrease the desire for alcohol, was developed in Europe and has been available in the USA since 2004. Acamprosate appears to reduce the long-lasting neuronal hyperexcitability that follows chronic alcohol use (
Kranzler & Gage 2008). The mechanisms are unclear but may include alterations in glutamate receptor gene expression. This medication suppresses the intake of alcohol in rats and, as with naltrexone, activity in the animal model predicts clinical efficacy. In double-blind studies, acamprosate has been shown to increase the likelihood of continuous abstinence in alcoholics and to shorten the period of drinking if the patient slips and consumes some alcohol.
The opiate receptor antagonist naltrexone has been reported in several clinical trials to reduce alcohol craving (
O'Brien 2005) as well as alcohol reward. Human laboratory studies of alcohol priming in non-treatment-seeking alcoholics demonstrated a reduction in alcohol craving and alcohol drinking in spite of the alcohol priming drink in participants who were not seeking treatment (
O'Malley et al. 2002).
Preclinical data have also shown an effect of alcohol on serotonergic systems. This has motivated trials using medications affecting that system. Ritanserin, a 5-hydroxytryptamine-2 (5-HT
2) receptor antagonist, was found to be no more effective than placebo in the treatment of alcoholism (
Johnson et al. 1996). Ondansetron, a 5-HT
3 antagonist, was found to reduce drinking in early-onset alcoholics both alone (
Johnson et al. 2000) and in combination with naltrexone (
Ait-Daoud et al. 2001). Specific craving studies have not been addressed with the serotonergic medications.