Overwhelming evidence confirms that prolonged stress adversely affects physical and behavioral parameters relevant to survival (
1). Because a wide variety of stressors reliably activate the hypothalamic-pituitary-adrenal (HPA) axis, and because glucocorticoids are the end product of HPA axis activation, these hormones have been most commonly seen as the
agents provocateurs, or even—in extreme cases—as the physical embodiment, of stress-induced pathology. Indeed, it has been suggested that prolonged overproduction of glucocorticoids, whether as a result of ongoing stress or a genetic predisposition to HPA axis hyperactivity, damages brain structures (especially the hippocampus) essential for HPA axis restraint (
2). Such damage, in turn, has been hypothesized to lead to a feed-forward circuit in which ongoing stressors drive glucocorticoid overproduction indefinitely (the “glucocorticoid cascade hypothesis”). Because of the capacity of high concentrations of glucocorticoids to disrupt cellular functioning in ways that can lead to a host of ills, this glucocorticoid overproduction is believed to contribute directly to many of the adverse behavioral and physiological sequelae associated with chronic stress (
2,
3).
Despite the popularity of the glucocorticoid cascade hypothesis, however, increasing data provide evidence that, in addition to glucocorticoid excess, insufficient glucocorticoid signaling may play a significant role in the development and expression of pathology in stress-related disorders. Indeed, since Hans Selye's initial definition of the stress response in the 1930s (
4), it has been recognized that under conditions of acute threat, glucocorticoids promote survival by mobilizing and directing bodily resources (
1,
3,
5). In addition to providing short-term adaptive advantages, it is becoming increasingly appreciated that glucocorticoids also confer longer-term stress-related benefits by shaping and eventually restraining stress-related physiological processes, including early, innate immune responses (inflammation), activation of the sympathetic nervous system (SNS), and stimulation of corticotropin releasing hormone (CRH) pathways, all of which are capable of producing a host of adverse health outcomes if allowed to continue unabated after crisis resolution (
5,
6). In contradistinction to concepts that privilege the pathological potential of glucocorticoids, this perspective suggests that because glucocorticoids constrain the very processes to which they initially contribute, conditions characterized by prolonged activation of the stress response might be especially prone to produce illness under conditions in which glucocorticoid signaling is insufficient.
If such an insufficiency of glucocorticoid signaling contributes to stress-related morbidity, one would expect to find evidence for its existence across a wide range of stress-related behavioral and physical disorders. It is the burden of this article to review the evidence for glucocorticoid insufficiency in these diseases. In the process, we will examine the possibility that unrestrained responsiveness of systems under glucocorticoid control, especially immune activation/inflammation, may, in turn, contribute to pathologies classically attributed to glucocorticoids.
We define insufficient glucocorticoid signaling as any state in which the signaling capacity of glucocorticoids is inadequate to restrain relevant stress-responsive systems, either as a result of decreased hormone bioavailability (e.g., hypocortisolism) or as a result of attenuated glucocorticoid responsiveness (e.g., secondary to reduced glucocorticoid receptor sensitivity). Thus defined, insufficient glucocorticoid signaling implies no specific mechanism or absolute deficiency but focuses instead on the end point of glucocorticoid activity. The fundamental question is whether the glucocorticoid message is getting through in a manner adequate to the environment (external and internal) in which an organism finds itself. Therefore, even in the case of glucocorticoid hypersecretion, glucocorticoid insufficiency can exist, if reduced glucocorticoid sensitivity in relevant target tissues outweighs excess circulating hormone.
HPA AXIS ORGANIZATION AND FUNCTION: PATHWAYS OF GLUCOCORTICOID SIGNALING
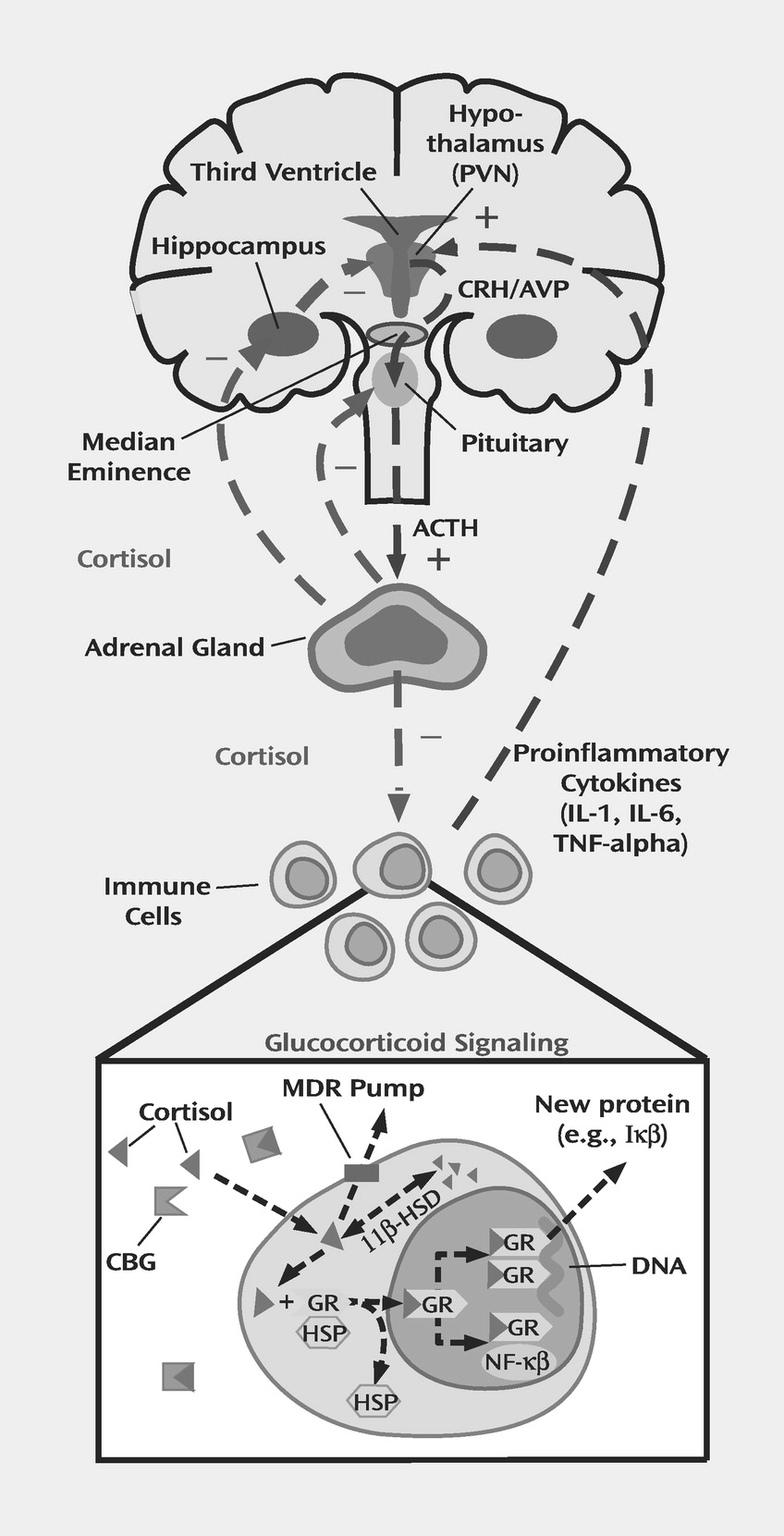
Figure 1 presents a simplified diagram of the HPA axis. Stressors of all sorts, both physical and psychological, activate the production and release of CRH from the paraventricular nucleus of the hypothalamus. Acting via the portal circulation, CRH, in conjunction with arginine vasopressin, induces the pituitary to produce adrenocorticotropic hormone (ACTH), which enters the bloodstream and causes the adrenal glands to release glucocorticoids (cortisol in humans and other primates, corticosterone in mice and rats) (
7). When produced in response to stress, glucocorticoids have myriad actions on the body that are primarily mediated via intracellular receptors. In their inactive state, these receptors exist in the cytosol within a topologically complex assembly of heat shock proteins that serve to stabilize the unbound receptor (
Figure 1) (
8). Glucocorticoids passively diffuse through the cellular membrane and bind to these receptors, a process which in turn promotes translocation to the nucleus. Within the nucleus, these ligand-activated receptors then either interact with other transcription factors or bind to specific DNA response elements with a resultant up- or down-regulation in the expression of various genes (
8).
Two distinct, but functionally related, intracellular receptors for glucocorticoids have been identified (
8,
9). Mineralocorticoid receptors have a high affinity for naturally occurring glucocorticoids, such as cortisol and corticosterone, as well as the salt-regulating hormone aldosterone. In contrast, glucocorticoid receptors bind avidly to synthetic steroids (such as dexamethasone) but have significantly lower affinity for endogenous hormones (cortisol and corticosterone). On the basis of these affinity differences, it is believed that mineralocorticoid receptors play a primary role in mediating glucocorticoid effects under basal conditions when hormone levels are low (
8,
9). As glucocorticoid levels rise, either in response to stress or as a function of the HPA circadian cycle, mineralocorticoid receptors saturate, and glucocorticoid receptors be come the chief transducer of glucocorticoid activity and are thus the primary mediators of feedback inhibition of CRH (and the HPA axis) (
8,
9). Because glucocorticoid receptors avidly bind dexamethasone and because most studies of HPA feedback integrity have used this synthetic glucocorticoid, e.g., in the dexamethasone suppression test (DST), far more information is available on the relationship between glucocorticoid receptor functioning and stress-related psychopathology.
When examined in the simplest terms, insufficient glucocorticoid signaling could result from decreased hormone bioavailability or decreased receptor-mediated signal transduction (which would translate into reduced hormone responsiveness). Within the HPA axis, a decrease in hormone bioavailability might stem from decreased production of upstream glucocorticoid secretagogues (CRH and ACTH), as well as from a primary deficit in adrenal hormone production and/or release. Decreased glucocorticoid bioavailability might also result from alterations in 1) binding proteins, which have been identified for both ortisol and CRH (
10), 2) enzymes such as 11-β-hydroxysteroid dehydrogenase, which metabolize endogenous glucocorticoid hormones upon entry into the cell (
11), and 3) the multidrug resistance pump, which extrudes cortisol but not corticosterone from the cell (
12). As with the production of hormones, decreased receptor-mediated signal transduction might occur as a result of abnormalities at any level of the HPA axis, including CRH and ACTH receptors, in addition to glucocorticoid and mineralocorticoid receptors. Receptor abnormalities might take the form of any combination of alterations in number, binding affinity, or functional capacity. Finally, it is important to note that any particular HPA axis abnormality may be a primary or a secondary adaptation to alterations elsewhere in the axis.
NEUROENDOCRINE CHANGES RELEVANT TO INSUFFICIENT GLUCOCORTICOID SIGNALING
It has been suggested that a decrease in upstream secretagogues, notably CRH, may lead to inadequate glucocorticoid signaling in several neuropsychiatric disorders, including atypical depression (
13), fibromyalgia (
14), and chronic fatigue syndrome (
15). Nevertheless, the most commonly reported neuroendocrine changes that might contribute to insufficient glucocorticoid signaling are reduced production and/or release of glucocorticoids (hypocortisolism) and reduced glucocorticoid responsiveness.
Hypocortisolism
As early as the 1960s, researchers noted that medically healthy people living in conditions of chronic stress frequently exhibited lower urinary and plasma cortisol concentrations than matched comparison subjects, with cortisol concentrations decreasing further during periods of heightened stress (
16). Decreased plasma, salivary, and urinary cortisol concentrations have also been reported in subjects suffering from posttraumatic stress disorder (PTSD), whether the syndrome developed as a result of combat exposure or a natural disaster, such as an earthquake, or following sexual and/or physical abuse in childhood (
17). Of note, several studies indicate that individuals who demonstrate low cortisol levels after an acute trauma, such as a motor vehicle accident, are at higher risk of subsequently developing PTSD symptoms (
18). Relevant to this finding is the observation that patients who receive stress-level doses of hydrocortisone as part of their treatment while in a medical intensive care unit (ICU) have lower rates of ICU-related PTSD symptoms (
19). Taken together, these data support the notion that glucocorticoids protect against the development of certain stress-related disorders.
Work by our group (
20,
21) has shown that women with a history of early life stress (childhood abuse), without current major depression, also exhibit reduced basal plasma cortisol concentrations and produce less cortisol in response to an ACTH challenge compared to women without a history of childhood abuse. Interestingly, these women exhibit exaggerated ACTH responses to either a psychosocial stressor or CRH, suggesting that hypocortisolism in these subjects represents an end organ (adrenal gland) adaptation to a sensitized (exaggerated) response to stress at the level of the pituitary and/or the hypothalamus (CRH).
Chronic disorders characterized by fatigue and pain, including chronic fatigue syndrome and fibromyalgia, may also be accompanied by reduced production and/or release of glucocorticoids (
22,
23). It is interesting that these disorders have been associated with high rates of trauma and chronic victimization, as well as PTSD symptoms (
24,
25). Patients with chronic fatigue syndrome have been found to exhibit decreased 24-hour urinary and plasma cortisol concentrations and decreased adrenal responses to maximal- and medium-dose ACTH stimulation (
23). Fibromyalgia has likewise been associated with decreased urinary cortisol levels and with decreased cortisol production after CRH challenge or a brief physical stressor (
22,
26). Reduced morning cortisol levels also have been reported in idiopathic pain syndromes, and decreased cortisol production in response to CRH has been found in patients with chronic pelvic pain (
24).
Reduced responsiveness to glucocorticoids
Reduced glucocorticoid responsiveness represents another major pathway that may contribute to insufficient glucocorticoid signaling. Perhaps the most compelling example of a stress-related disorder with evidence of reduced glucocorticoid responsiveness is major depression. Numerous studies over the past four decades have repeatedly shown that, as a group, patients with major depression have reduced responses to glucocorticoids as assessed both in vivo and in vitro (
8,
27). The DST and its new and improved version, the dexamethasone-CRH stimulation test, are the two in vivo tests that have received the most attention. Rates of impaired glucocorticoid responsiveness during major depression (as assessed by nonsuppression of cortisol on the DST or dexamethasone-CRH test) vary from approximately 25% to 80% (
27), depending on depressive symptoms (the highest rates are found for melancholic, or endogenous, subtypes) (
28), age (older subjects are more likely to exhibit nonsuppression), and the technique used for assessment (the dexamethasone-CRH test is more sensitive than the DST) (
27,
29). Of note, both the DST and the dexamethasone-CRH test have been shown to powerfully predict clinical response (
30,
31), and in the case of the dexamethasone-CRH test, there is evidence that impaired glucocorticoid responsiveness represents a genetically based risk factor for the development of depression (
32).
Complementing in vivo findings, results of in vitro studies have demonstrated that peripheral immune cells from patients with major depression exhibit decreased sensitivity to the well-known immunosuppressive effects of glucocorticoids (
8). Whereas normal subjects show a marked inhibition of in vitro natural killer (NK) cell activity, lymphocyte proliferation, or cytokine production after exposure to a glucocorticoid (usually dexamethasone), patients with major depression, especially DST nonsuppressors, consistently show an attenuated inhibitory response (
33–
36). It should be noted that in vitro studies obviate concerns that abnormalities in the DST and dexamethasone-CRH test solely reflect reduced dexamethasone bioavailability (secondary to increased dexamethasone clearance) or increased hypothalamic drive.
Since dexamethasone binds avidly to the glucocorticoid receptor, dexamethasone nonsuppression during major depression provides evidence of impaired glucocorticoid receptor signaling. Consistent with this notion, transgenic mice whose glucocorticoid receptor has been genetically reduced by 30%–50% require 10-fold higher doses of dexamethasone to suppress both basal and CRH-stimulated corticosterone release (
37). However, we know of no data demonstrating that the structural integrity of the glucocorticoid receptor itself is altered in depressed patients (
8,
27). Nevertheless, several lines of evidence suggest that alterations in neurotransmitter-linked signal transduction pathways that regulate glucocorticoid receptor function may contribute to diminished glucocorticoid receptor signaling in major depression (
8,
27). Indeed, altered glucocorticoid receptor signaling has been proposed as a major factor in the pathogenesis of the disorder (
8,
27).
GLUCOCORTICOID TONE IN STRESS-RELATED DISORDERS: IS THE MESSAGE GETTING THROUGH?
We have provided evidence that stress-related disorders are characterized by neuroendocrine changes that bespeak insufficient glucocorticoid signaling. However, decreased hormone production (e.g., hypocortisolism) or decreased glucocorticoid responsiveness (e.g., dexamethasone nonsuppression) can be counterbalanced by adaptive changes that attempt to normalize glucocorticoid signaling. Consistent with this notion, Yehuda and colleagues have demonstrated up-regulation of glucocorticoid receptors and increased sensitivity to dexamethasone in patients with PTSD (who exhibit hypocortisolism) (
17). Moreover, glucocorticoid hypersecretion is a common and reliable finding in patients with major depression (
8), occurring in conjunction with reduced glucocorticoid responsiveness (dexamethasone nonsuppression) approximately 60% of the time (
38).
One approach to resolving the question of whether too much or too little glucocorticoid signal ultimately “gets through” is to examine target tissues or systems whose function is regulated in part by glucocorticoids. In effect, such tissues and/or systems provide a naturalistic assay to evaluate whether overall neuroendocrine changes are more consistent with an excess or insufficiency of glucocorticoid tone. A related approach is to examine pathologies that typify stress-related disorders and determine whether these pathologies are consistent with glucocorticoid excess or insufficiency. Finally, treatments that are known to be effective for stress-related disorders (i.e., antidepressants) can be evaluated in terms of their impact on glucocorticoid signaling pathways. Each of these approaches will be considered.
Target systems regulated by glucocorticoids
Immune system.
A critical function of glucocorticoids is to mobilize and shape immune responses during stress (
6). Virtually all stressors, including infections, physical trauma, and even psychological insults, are associated with immune activation and the release of proinflammatory cytokines such as tumor necrosis factor alpha (TNF alpha) and interleukin-1 (IL-1) and interleukin-6 (IL-6) (
39). Through their inhibitory effects on nuclear factor κβ signaling pathways, glucocorticoids are the most potent anti-inflammatory hormones in the body and thereby serve to suppress the production and activity of proinflammatory cytokines during stressor exposure and return the organism back to baseline (homeostasis) after cessation of the stressor (
6,
40,
41). Indeed, in the absence of glucocorticoids, animals quickly succumb to the ravages of the inflammatory response (
41).
Consistent with inadequate glucocorticoid-mediated feedback inhibition of immune responses, evidence of immune activation (early, innate immune responses, in particular) has been reported in a number of stress-related neuropsychiatric disorders. Best characterized in this regard is major depression. The inflammatory changes reported include increased levels of acute-phase reactants, increased plasma and CNS concentrations of proinflammatory cytokines (especially IL-1 in CNS and IL-6 inplasma), increased levels of prostaglandins, and an increase in stimulated in vitro peripheral blood monocyte production of proinflammatory cytokines (
42). Of note, increased concentrations of plasma IL-6 have been found to positively correlate with post-DST cortisol levels (
36), suggesting that DST nonsuppression identifies a group of depressed subjects most likely to demonstrate immune activation. In addition, depression has been shown to predict exacerbations in diseases with an inflammatory component, including both rheumatoid arthritis (
43) and multiple sclerosis (
44).
Immune activation has also been reported in stress-related disorders characterized by decreased cortisol production (i.e., hypocortisolism), supporting the contention that overall glucocorticoid signaling capacity, or tone, is decreased in these disorders, despite evidence for increased receptor sensitivity (in PTSD). These disorders include PTSD, chronic fatigue syndrome, and fibromyalgia. For example, serum concentrations of IL-1-β and CSF levels of IL-6 have been found to be elevated in patients with combat-induced PTSD (
45), and serum concentrations of both IL-6 and its soluble receptor have been found to be elevated in patients with PTSD secondary to civilian trauma (
46). Patients with chronic fatigue syndrome and fibromyalgia have also exhibited immune activation, as evidenced by increased plasma concentrations of acute phase reactants and increased plasma concentrations, and/or peripheral blood mononuclear cell production, of proinflammatory cytokines, including IL-6, TNF-alpha, and IL-1 (
47–
50).
Unlike immune activation, which is consistent with glucocorticoid insufficiency, the impairments in T cell proliferation and NK cell activity frequently reported in stress-related disorders (especially major depression) might be accounted for by either excessive or insufficient glucocorticoid signaling. Glucocorticoids are well known to suppress multiple aspects of lymphocyte function; however, several studies of patients with major depression have failed to show a relationship between altered T cell responses or NK cell activity and plasma or urinary concentrations of free cortisol (
34,
51). Proinflammatory cytokines are also capable of disrupting T cell signaling pathways and inhibiting NK cell activity (
52,
53), thereby providing a potential link between insufficient glucocorticoid signaling, immune activation, and reduced lymphocyte responsiveness. Indeed, mice with impaired glucocorticoid receptor function exhibit reduced T cell responses secondary to chronic activation of nitric oxide pathways (
54). Nevertheless, we know of no studies in which an attempt was made to correlate low T cell proliferation or NK cell activity with proinflammatory cytokine production in the context of major depression.
SNS.
Glucocorticoids play an important role in the regulation of the SNS. In addition to subserving permissive effects on relevant synthetic enzymes and receptors for catecholamines, endogenous glucocorticoids have been shown to restrain SNS responses under resting conditions and after stress (
55). Administration of high concentrations of glucocorticoids to laboratory animals has been found to inhibit, enhance, or have no effect on SNS activity (
55,
56). In humans, however, high concentrations of glucocorticoids are more reliably associated with reduced SNS responses (
57–
59). For example, short-term (1-week) administration of glucocorticoids to normal volunteers has been shown to reduce sympathetic nerve activity and diminish circulating concentrations of norepinephrine (
59). In addition, in Cushing's disease (a condition of chronic glucocorticoid exposure), a negative relationship has been found between concentrations of urinary free cortisol and plasma norepinephrine, both under resting conditions and after orthostatic challenge (
58).
Consistent with evidence of glucocorticoid insufficiency in stress-related disorders is the association of both major depression and PTSD with increased CSF and plasma concentrations of catecholamines and their metabolites (
60–
62). Moreover, in contrast to patients with Cushing's disease, patients with major depression have been found to exhibit a positive relationship between postdexamethasone concentrations of cortisol and plasma catecholamine concentrations, further suggesting that cortisol elevations following dexamethasone reflect insufficient glucocorticoid signaling in this disorder (
63). In patients who develop PTSD, reduced cortisol levels at the time of trauma have been posited to contribute to exaggerated catecholaminergic responses and increased arousal and anxiety symptoms (
64,
65). Finally, exaggerated SNS responses may contribute to increased proinflammatory cytokine production that exacerbates pathology in peripheral tissues, including the heart (
66).
CRH.
As indicated in
Figure 1, glucocorticoids are well known to inhibit CRH activity in the paraventricular nucleus of the hypothalamus (
7). Indeed, postmortem samples from patients treated with glucocorticoids exhibit marked reductions in the expression of CRH in hypothalamic neurons (
67). Consistent with insufficient glucocorticoid regulation of CRH function in stress-related disorders, hyperactivity of CRH pathways has been observed in PTSD, as well as major depression (despite increased levels of circulating glucocorticoids). For example, investigators have found increased CRH mRNA and protein levels in the paraventricular nucleus of postmortem brain samples from patients with major depression (
68). Moreover, depressed patients have been shown to exhibit increased concentrations of CRH in CSF (
69). Similar changes in CRH activity, including increased CSF CRH concentrations, have been described for patients with PTSD (
70). Because CRH has behavioral effects in animals that include alterations in activity, appetite, and sleep, it has been suggested that CRH hypersecretion in stress-related diseases might contribute to the behavioral alterations in these disorders (
7).
Stress-related pathologies and the role of glucocorticoids
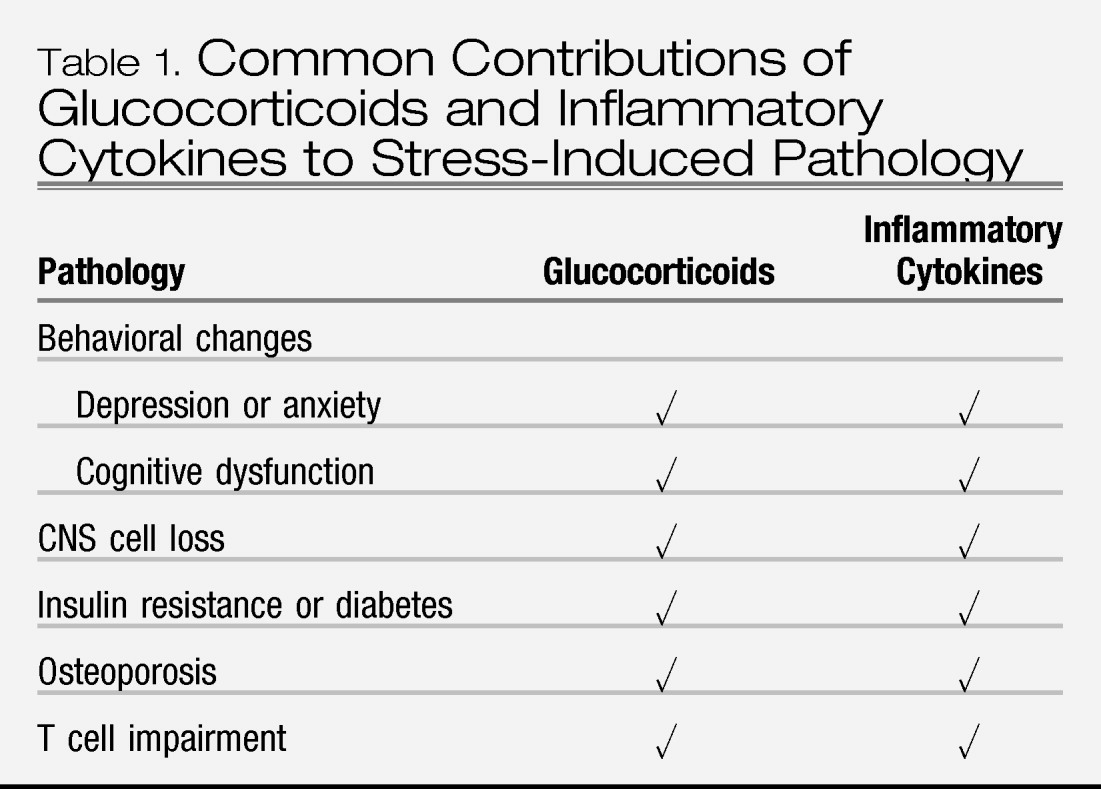
A number of pathologies associated with stress have been attributed to glucocorticoid excess, including volumetric changes in the brain, insulin resistance and/or diabetes, and osteoporosis. However, in each case, the data are correlative in nature, and we know of no studies that have directly tested the role of glucocorticoids (e.g., by antagonizing glucocorticoids and reversing the pathological changes). Moreover, each of these pathologies can be caused by consequences of insufficient glucocorticoid signaling, especially immune activation and inflammation (
Table 1).
Volumetric changes in the brain.
A central tenet of the glucocorticoid cascade hypothesis is that glucocorticoid excess results in damage to key brain structures involved in HPA axis restraint, including, most notably, the hippocampus (
2). Consistent with this hypothesis, volume reductions in the hippocampus and other brain regions, as determined by magnetic resonance imaging, have been described in both PTSD (
71) and major depression (
72). However, postmortem brain specimens from depressed patients have failed to reveal significant pathology in the hippocampus at the cellular level, including hippocampal areas at putative high risk for glucocorticoid-mediated damage (
73,
74). Moreover, work in rhesus monkeys suggests that glucocorticoid receptor expression in primates may dramatically differ from that in rats (
75), the species from which the glucocorticoid cascade hypothesis was initially derived.
Unlike the hippocampus, the frontal cortex of patients with major depression has been found to exhibit neuropathologic abnormalities, including loss of neuronal elements, especially glia (
76). These changes could result from either glucocorticoid excess or insufficiency. In support of processes linked to insufficient glucocorticoid signaling, unrestrained inflammation and increased release of proinflammatory cytokines have been shown to influence cell survival in the CNS (
77,
78). For example, inflammatory cytokines (especially IL-1) are well known to contribute to cell death (neurons and glia) after multiple types of CNS trauma (
77). Even in the absence of trauma, administration of IL-1 in the striatum has been shown to lead to widespread cell loss in the frontal cortex when coadministered with glutamate agonists (
77). Similarly, TNF alpha is neurotoxic to primary cultures of septohippocampal cells and leads to apoptotic death in primary cortical neurons (
77). Moreover, recent data (
78) indicate that rodents treated with the antiglucocorticoid RU486 exhibit dramatic increases in TNF-alpha-mediated neurodegeneration that is dependent on activation of neurotoxic pathways involving both nitric oxide and caspase. These data emphasize the fundamental role played by glucocorticoids in protecting the brain during immune activation/inflammation (
78). Finally, it should be noted that unrestrained release of CRH secondary to insufficient glucocorticoid-mediated feedback inhibition may also contribute to neuronal degeneration (
79).
Insulin resistance and diabetes.
Given the well-known capacity of glucocorticoids to influence glucose metabolism, a link between stress-related disorders, glucocorticoids, and insulin resistance (with its attendant hyperglycemia, dyslipidemia, hypertension, and abdominal obesity) has been proposed, although not established (
80). For example, patients with major depression have been noted to exhibit insulin intolerance (
81), and major depression has been associated with a worse outcome in diabetic patients (
82). However, we know of no study that has directly linked these findings to glucocorticoid excess (
83). Moreover, although patients with major depression have been shown to exhibit a greater waist-to-hip ratio (an indicator of abdominal obesity), a relationship between greater waist-to-hip ratio and excessive glucocorticoid activity in major depression has not been demonstrated as far as we know (
84–
86). Of note, in studies on individuals without psychiatric disorders, a higher body mass index and waist-to-hip ratio have been associated with both increased and decreased secretion of cortisol (
87). Moreover, in a well-controlled study on middle-aged men with a high body mass index, men with a higher waist-to-hip ratio exhibited reduced, rather than enhanced, glucocorticoid responses after exposure to either a laboratory stressor or CRH; this finding is consistent with evidence of insufficient glucocorticoid signaling (
88).
As a result of insufficient glucocorticoid signaling, release of inflammatory elements from glucocorticoid-mediated inhibitory control may contribute to altered glucose metabolism in stress-related disorders. A number of studies have demonstrated that proinflammatory cytokines, including TNF-alpha and IL-6, are associated with insulin resistance, diabetes, and obesity (
89–
91). Moreover, patients with inflammatory disorders, including rheumatoid arthritis, exhibit increased rates of insulin resistance (
92). The mechanism of these effects is believed to be related in part to TNF-alpha-mediated inhibition of insulin receptor tyrosine kinase activity, as well as inhibition of genes required for insulin signaling and glucose transport (
93). A recent study in rodents indicates that obesity- and diet-induced insulin resistance, along with elevated levels of triglycerides and free fatty acids, can be reversed by treatment with anti-inflammatory agents or genetic disruption of inflammatory signaling pathways (
94). Of special relevance to the role of glucocorticoid signaling are data indicating that fat cell production of IL-6, a major determinant of circulating IL-6 levels in obese subjects, is negatively regulated by glucocorticoids (
95).
Osteoporosis.
Stress-related disorders—especially major depression—have been associated with osteoporosis (
96,
97). Given the inhibitory effects of glucocorticoids on bone metabolism, osteoporosis has been considered evidence of increased glucocorticoid activity in stress-related disorders (
97). Nevertheless, limited data are available that correlate cortisol levels with bone mineralization. Although one study (
98) showed an inverse relationship between plasma cortisol level and bone mineral density in depressed patients, at least two studies (
96,
99) have failed to replicate this finding.
It should be noted, however, that pathways linked to insufficient glucocorticoid signaling and unrestrained inflammation could also contribute to the development of osteoporosis. For example, the proinflammatory cytokine IL-6 (levels of which are elevated in major depression and PTSD) is a potent antagonist of bone formation and is believed to be one of the major mediators of bone loss in postmenopausal women (
100). However, as is the case with cortisol, studies have not established a clear link between plasma concentrations of proinflammatory cytokines and depression-related bone loss (
96).
Behavioral alterations.
Given evidence for an association between glucocorticoid insufficiency and immune activation, the question arises as to whether proinflammatory cytokines can contribute to neuropsychiatric and physical symptoms associated with stress-related disorders, including symptoms traditionally ascribed to glucocorticoids. In this regard, proinflammatory cytokines have been shown to induce a syndrome of “sickness behavior” (
101). This syndrome, which includes anhedonia, anorexia, fatigue, sleep alterations, and cognitive dysfunction, has many features that overlap with stress-related neuropsychiatric and physical disorders, including major depression, chronic fatigue syndrome, fibromyalgia, and PTSD.
Both cytokines and their receptors are found in brain regions that are centrally involved in the mediation of emotion and behavior, such as the hypothalamus and hippocampus (
102). Blocking these cytokines has been shown to attenuate or abolish symptoms of sickness behavior after infection or cytokine administration in laboratory animals (
103). Relevant to the pathophysiology of behavioral changes in stress-related disorders are the findings that proinflammatory cytokines are potent stimulators of CRH in multiple brain regions and that they influence monoamine neurotransmitter turnover in the hypothalamus and hippocampus (
39,
101). Proinflammatory cytokines can also lead to hyperalgesia and have been implicated as major contributors to chronic pain symptoms, which commonly accompany stress-related disorders (
104).
Glucocorticoid signaling.
Several lines of evidence suggest that cytokines may also contribute directly to insufficient glucocorticoid signaling by diminishing glucocorticoid receptor functional capacity. Administration of cytokines in vivo and in vitro has been shown to reduce both glucocorticoid receptor number and function (
105,
106). Mechanisms by which cytokines may alter glucocorticoid receptor activity include inhibition of glucocorticoid receptor translocation from cytoplasm to nucleus (
106), induction of inert isoforms of the glucocorticoid receptor (glucocorticoid receptor β) (
107), and activation of mitogen-activated protein kinase signaling pathways that in turn inhibit glucocorticoid receptor activity (
105,
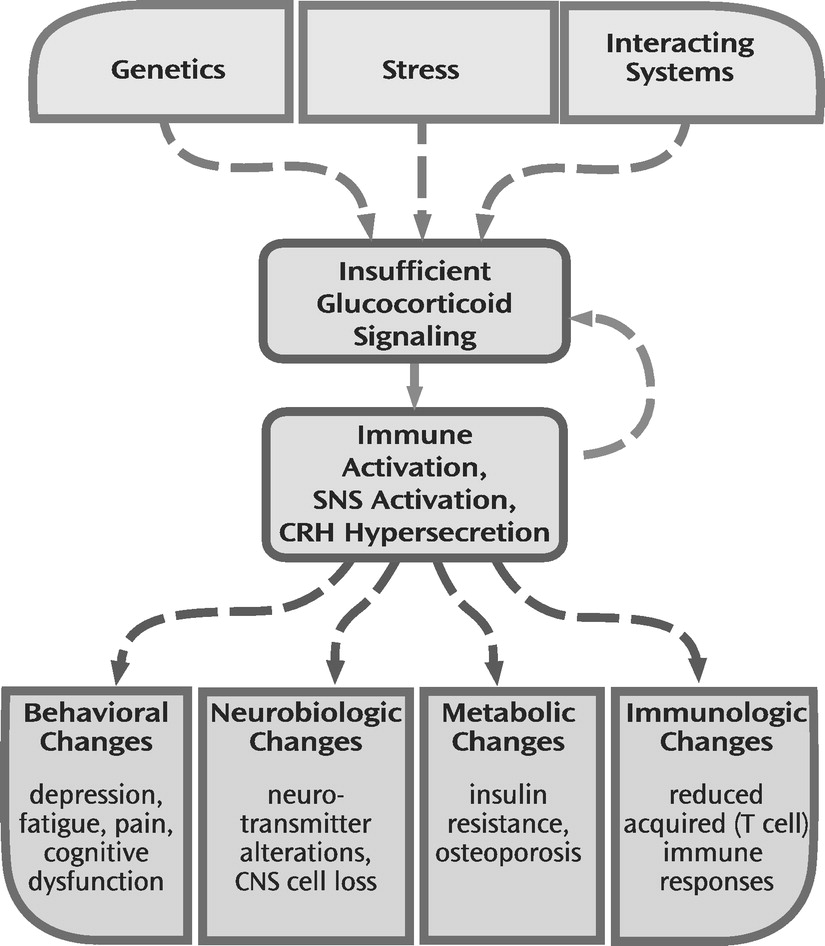
106). These findings make plausible a scenario in which glucocorticoid insufficiency contributes to unrestrained cytokine release, which in turn further impairs glucocorticoid receptor functioning, leading to a feed-forward inflammatory cascade (
Figure 2).
Effects of antidepressants on glucocorticoid signaling
Antidepressants are a front-line treatment for stress-related disorders, including both PTSD and major depression. Successful antidepressant treatment is associated with normalization of altered glucocorticoid-mediated inhibitory feedback in patients with major depression, as assessed by either the DST or dexamethasone-CRH stimulation test (
8,
27,
31). Similarly, long-term antidepressant treatment for patients with major depression restores appropriate glucocorticoid inhibitory control of immune cell function, as assessed in vitro (
8,
27).
In support of the notion that antidepressant effects on neuroendocrine function are related to enhancement of glucocorticoid signaling, a number of animal studies have demonstrated that long-term antidepressant administration increases both the number and functional capacity of corticosteroid receptors in brain regions known to be of key importance in HPA axis regulation, including the hippocampus and hypothalamus (
8,
27). This effect has been observed most consistently with tricyclic antidepressants, although other agents and somatic treatments have been shown to increase glucocorticoid receptor expression and/or function in the brain (
8,
27). Preclinical data also demonstrate that long-term antidepressant administration reverses HPA axis alterations that result from glucocorticoid receptor dysfunction (
8,
27). For example, in transgenic mice with diminished CNS glucocorticoid receptors, long-term treatment with amitriptyline has been shown to reverse baseline DST nonsuppression, while at the same time correcting species-typical behavioral abnormalities that are reminiscent of those seen in human depression (
27).
Regarding the cellular mechanisms of these effects, in vitro experiments have demonstrated that antidepressants are capable of translocating the glucocorticoid receptor from cytoplasm to nucleus, even in the absence of glucocorticoids (
108). Antidepressants also enhance dexamethasone-induced gene transcription mediated by the glucocorticoid receptor (
8,
27,
108). These antidepressant effects may be related to the impact of these drugs on second messenger pathways involved in glucocorticoid receptor regulation, including activation of the cyclic adenosine monophosphate/protein kinase A (cAMP/PKA) cascade (
109). Activation of both cAMP and PKA has been shown, in turn, to enhance glucocorticoid receptor functioning (
8,
27,
110,
111).
Finally, glucocorticoid agonists and antagonists (including glucocorticoid synthesis inhibitors) have been used to treat stress-related neuropsychiatric disorders, in particular major depression. Indeed, on the basis of theories that excessive glucocorticoid activity plays an integral role in the pathophysiology of major depression, several clinical studies using glucocorticoid antagonists, including RU486, are under way. Both types of treatments (glucocorticoid agonists and antagonists) have shown some efficacy in relatively limited clinical trials (
112,
113). Nevertheless, on the basis of these data alone, it is difficult to conclude whether the results provide support for the pathophysiologic relevance of excessive and/or insufficient glucocorticoid signaling.
GLUCOCORTICOID INSUFFICIENCY AND ADAPTATION: IN SEARCH OF EVOLUTIONARY CAUSATION
In evaluating the hypothesis of glucocorticoid insufficiency, it is of interest to explore potential adaptive reasons for development and maintenance of pathways for insufficient glucocorticoid signaling in the mammalian gene pool.
Although autoimmunity remains an ongoing risk whenever the immune system is activated, prolonged or repeated exposure to immune stimuli might predispose an individual to reduced glucocorticoid signaling as a means of freeing bodily defenses from inhibitory control in the face of an ongoing infectious threat. Such a release of inflammatory processes might be adaptive under conditions in which recurrent infection is likely and immune readiness is an attendant requirement.
On the basis of recent work using a social disruption paradigm in rodents, Avitsur and colleagues (
114) have proposed an evolutionary explanation for stress-related reduced responsiveness to glucocorticoids (glucocorticoid resistance). These investigators observed that defeated, but not victorious, rats demonstrated decreased immune system sensitivity to glucocorticoid-mediated inhibition. Closer examination revealed that development of glucocorticoid resistance correlated with assumption of a subordinate behavioral profile after defeat and with the number of wounds received in fighting with aggressive intruder mice. Avitsur et al. proposed that because the submissive behavioral profile is associated with more wounds, the development of glucocorticoid resistance may be an adaptive mechanism that allows inflammatory healing to occur in the context of stress-related increases in glucocorticoids. In this regard, it is interesting to speculate that across phylogenetic development, the risk of being wounded by conspecifics in hierarchical groups may have led to a condition for subordinate animals in which overall survival was favored by promotion of rapid, nonspecific immunity at the expense of more slowly developing specific immunity. Such rapidly deployed nonspecific, or innate, immunity is favored by proinflammatory cytokines, which also (especially TNF-alpha) paradoxically suppress the development of specific (acquired) immune responses, including, most notably, T cell responses such as T cell proliferation (suppressed in major depression) and T cell receptor signaling (
53).
In addition to effects on the immune system, reduced glucocorticoid signaling (whether achieved at the level of the hormone or its receptor) may also promote nervous system states that benefit successful adaptation to chronically stressful situations. For example, given the well established role of noradrenergic pathways in memory consolidation for emotionally laden experiences, enhanced noradrenergic function (as a result of reduced glucocorticoid-mediated restraint) might increase arousal and facilitate both memory formation and subsequent avoidance of potentially hostile environments (
65).
SUMMARY
Previous theories of stress-related pathologies have privileged the pathological potential of glucocorticoids. Herein we document that, in a number of instances, stress-related neuropsychiatric disorders may be characterized by insufficient glucocorticoid signaling as manifested by hypocortisolism or impaired glucocorticoid responsiveness associated with evidence of immune activation/inflammation, increased SNS tone, and hypersecretion of CRH. Hyperactivity of these stress-responsive systems, especially inflammation, in turn, may contribute to the behavioral features of these disorders, including depressed mood, anhedonia, fatigue, pain, and cognitive dysfunction, as well as the neurobiological, metabolic, and immunologic consequences of stress. On the basis of these data, the significance of stress-related insufficiency in glucocorticoid signaling, in terms of both adaptation and disease, is emphasized. Treatment implications center on approaches to enhancing glucocorticoid signaling pathways.