This paper proposes an elaboration of the current neurobiological theory of the memory mechanisms intrinsic to posttraumatic stress disorder (PTSD). It is not proposed as a final or fully comprehensive theory of the etiological mechanisms of this disorder. However, we believe that the theory represents a substantive advance because it incorporates clinical features of PTSD that currently are not adequately explained. We hope that the ideas presented may contribute to further theory development, with the ultimate goal of improving treatment.
LIMITATIONS OF THE CURRENT FEAR-CONDITIONING MODEL
Progress in understanding the biological basis of anxiety disorders has been achieved in the past decade largely by building on a preclinical model involving fear conditioning in animals. In particular, the current neurobiological model of PTSD derives by analogy from results of conditioning experiments, primarily in rodents.
5 In these investigations, a previously benign stimulus, such as an auditory tone, elicits autonomic fear responses after it has been paired with a highly aversive event, usually electric shock. Brain lesion, pharmacological, and electrophysiological studies of fear conditioning have identified the amygdaloid nuclei, complexes of neurons located in the temporal lobes, as central to the neuroanatomic circuitry responsible for the perception, expression, and memory of fear.
5–8The term “fear conditioning” refers to the acquisition of specific autonomic responses and is not dependent on, and may not involve, the conscious recall of experience. Experimental studies and clinical observations in neurologically impaired individuals have demonstrated that this form of conditioning may occur in the absence of conscious memory for aversive or catastrophic events.
9–11 Not surprisingly, the fear-conditioning model provides an effective account of the etiology of some symptoms of PTSD that are the result of memory intensification, particularly the arousal symptoms that result from the association of a neutral stimulus and an aversive one (classical conditioning).
Although the analogy between fear conditioning in animals and psychopathology in humans has played a critical role in understanding anxiety disorders, it has its limits with respect to PTSD, principally because it does not account for components of the clinical syndrome that involve alterations in episodic (autobiographical) memory. In particular, the model fails to incorporate two peritraumatic disturbances of conscious memory that are regular features of PTSD: impairment of memory for events immediately preceding and following the traumatic experience and the dissociation of the memory of the traumatic experience from ordinary autobiographical memory.
12,13 Both are reported consistently in clinical series.
14,15 In addition, both have been shown to be associated with increased risk of developing PTSD as a consequence of combat exposure,
16 motor vehicle and other accidents,
17,18 and assault.
19 Furthermore, memory impairment analogous to the peritraumatic memory impairment observed in PTSD is empirically reproducible in nonclinical human populations exposed to laboratory simulations of emotionally traumatic situations.
20,21Clearly, PTSD most appropriately is conceived as a clinical condition that involves both memory
intensification for the core traumatic event and memory
impairment for the context surrounding the trauma, which also comprises
dissociation of the experience from ordinary autobiographical memory. Because disturbances of episodic memory for peritraumatic events are not explained directly in the fear-conditioning model, it generally has been presumed that these conscious memory disturbances are secondary symptoms that develop as psychological defenses to suppress and otherwise avoid the primary symptoms of intensified memory or intrusive reexperience of the trauma. The latter describes the commonsense rationale for the occurrence of peritraumatic memory disturbance, which also is reflected in the criteria of the DSM,
1 in which peritraumatic memory impairment and dissociation are included among the avoidance cluster. A more adequate explanation of the coupling of trauma-induced intensification of memory and peritraumatic memory disturbance would allow amnesia and dissociation to be framed as brain-based disorders of episodic memory triggered by excessive emotional arousal, rather than, merely descriptively, as psychodynamic defenses against catastrophic fear. We will argue that these peritraumatic memory disturbances do reflect the operation of neurobiological processes fundamental to the etiology of PTSD rather than the effects of secondary protective psychological responses.
Certainly, the most intractable problem for the fear-conditioning model is the co-occurrence of memory intensification and amnesia, two apparently contradictory phenomena. This problem exists because in the fear-conditioning model PTSD has only one cause: intensification of memory for the traumatic experience as the result of conditioning.
5 Although proponents of the fear-conditioning model have acknowledged the need to incorporate a neurobiological explanation for peritraumatic memory impairment, to date there has been only one evident attempt to do so. LeDoux
3 speculated that peritraumatic amnesia might result from amygdalar activation of the hypothalamic-pituitary-adrenal axis, a neuroendocrinological mechanism that results in hippocampal dysfunction as a result of chronic stress.
22 However, as LeDoux himself indicated, the neuroendocrinological mechanism is improbable for a variety of reasons, among them the time scale required for such an effect.
3In view of both theoretical and clinical concerns, then, a neurobiological account of PTSD is required that incorporates the disturbances of conscious cognitive processes that are as fundamental to the disorder as disturbances of conditioned learning. A satisfactory neurobiological model of PTSD needs to explain the manner in which exposure to an extreme stimulus may result in both memory intensification and memory impairment and would suggest how these phenomena interact to produce the range of symptoms that constitute PTSD.
A REVISED MODEL OF PTSD
The model presented here accepts the fear-conditioning explanation for the noncognitive symptoms of PTSD and extends it to suggest a neurobiological explanation for the cognitive memory disturbances associated with traumatic experience. The model accounts for the cognitive memory disturbances of PTSD by postulating a coupled, two-component process mediated by the amygdala during exposure to emotionally intense stimulation. One component consists of processes that result in the intensification of memory for the traumatic stimulus. The other component consists of processes that result in diminution of conscious memory for stimuli temporally proximate to the traumatic stimulus.
The coupling of memory intensification and amnesia might appear to be a paradox that is unique to psychopathology. However, this is not the case. Indeed, our understanding is that PTSD is a special case of the normal operation of memory systems. Consequently, the proposed neurobiological model accounts for memory phenomena characteristic of PTSD as well as other memory phenomena, including results of laboratory investigations of emotional memory in nonclinical human populations. An example of these laboratory investigations emanates from a classic observation termed the
von Restorff effect, the augmentation of memory for one novel item in an otherwise neutral or homogeneous stimulus field.
23 One prominent finding of studies using the von Restorff paradigm is the co-occurrence of intensification of memory for a sufficiently arousing critical stimulus with memory impairment for temporally proximate neutral material.
Some of these studies have been explicit attempts to investigate eyewitness memory phenomena under controlled conditions. In one study,
20 subjects were shown one of two brief films used to train bank employees. An emotional version differed from a neutral version only by the inclusion of one scene depicting the shooting of a boy in the face. Retention of another specific detail common to both conditions was considerably poorer in the subjects who viewed the emotionally violent version in comparison to those who viewed the neutral film. Other experimental investigations of emotionally induced memory disturbances have investigated the amnestic effect on neutral material in controlled list learning experiments where the critical item is, for example, a photograph of a deformed child's face (versus a normal-appearing face in the control group) embedded within a sequence of drawings of various objects.
24Investigation of the variables associated with experimentally induced amnesia was developed by Tulving
25 and by Detterman and co-workers.
26–30 Depending on the intensity or duration of a distinctive stimulus in a list of items, the Detterman experiments established that enhanced recall for the distinctive stimulus can be coupled with impairment of memory for the proximate neutral stimuli in a manner that mimics the characteristics of neurological retrograde and anterograde amnesia. These experiments suggest the existence of a general neurobiological mechanism in which memory intensification and diminution are coupled, the phenomenon encountered in pathological degree in PTSD. The methodology of these experiments also suggests a means to investigate cognitive memory phenomena associated with PTSD in the absence of fear conditioning, opening the door to investigations in human subjects.
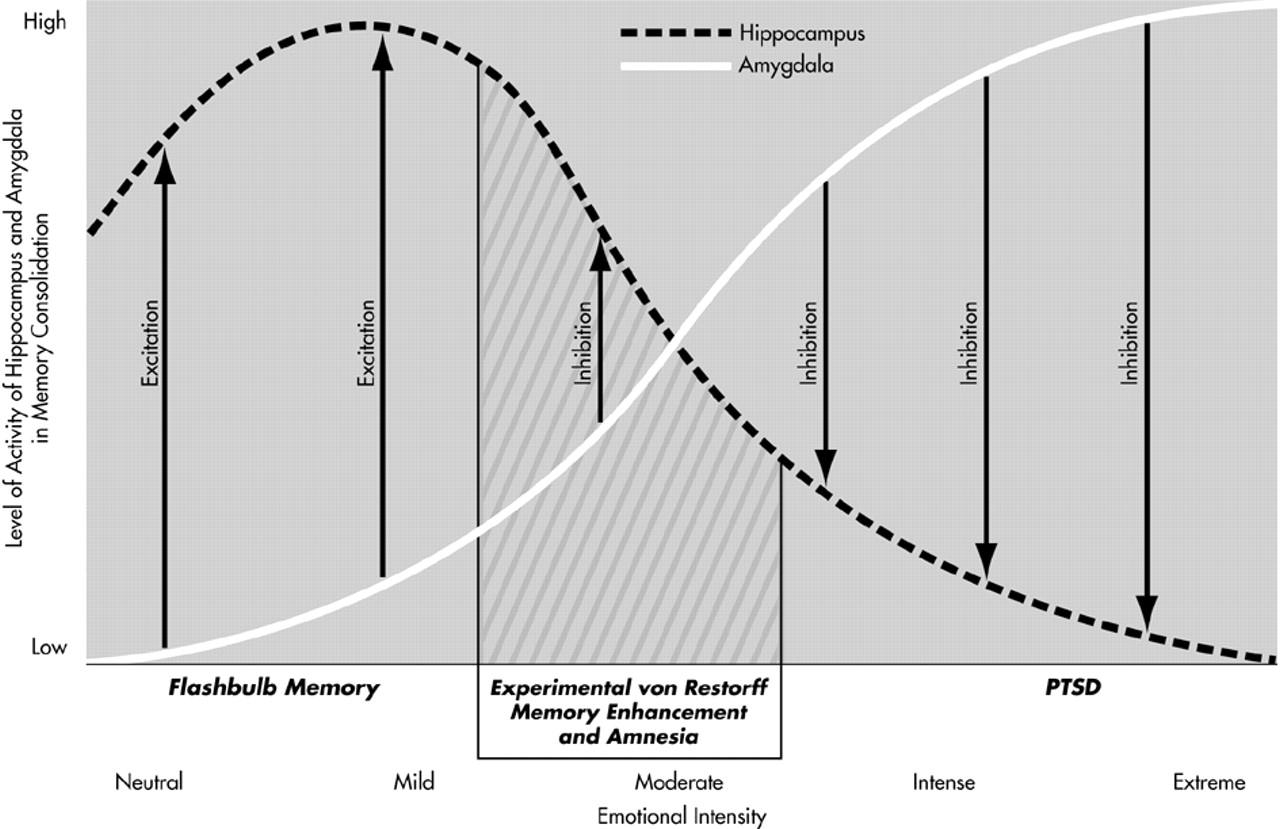
We propose that the processes underlying the coupling of memory intensification and memory decrement involve interaction of the amygdala and hippocampus in the context of sufficiently arousing stimuli, and we suggest a specific and testable neurobiological model to explain these effects. This model, represented diagrammatically in
Figure 1, is consistent with the anatomy and neurophysiology of the amygdala and hippocampus. With respect to the results of the von Restorff experiments, the model proposes that the critical item in the list is sufficiently arousing to engage the amygdala to 1) Consolidate memory for the critical item in the amygdala itself, and 2) suppress consolidation of the critical item by the hippocampus. As illustrated in
Figure 1, both effects occur in gradient fashion as a function of the level of arousal induced by the stimulus.
Most generally with respect to the relationship between emotional stimulation and neurophysiology, the model proposes that the amygdala is recruited increasingly as a function of emotional intensity. At modest levels of arousal, the amygdala potentiates hippocampal function and, accordingly, enhances memory. This phenomenon corresponds to the
flashbulb effect, the enhancement of episodic memory for events that are moderately emotionally arousing
31 (see
Figure 1). At greater levels of stimulus intensity, the amygdala begins to inhibit hippocampal consolidation of memory, both for the emotional stimulus itself and for proximate neutral stimuli. Beyond this threshold, as the amygdala increasingly is activated the hippocampus increasingly is suppressed and contextual features proximate to the emotionally intense stimulus will be increasingly less well consolidated in episodic memory.
The critical item in the von Restorff paradigm induces amygdalar inhibition of hippocampal consolidation not only for the critical item, but also for the neutral items as a function of their temporal proximity to the critical item. The temporal spread of inhibition around the critical item results in anterograde and retrograde amnesia. Because the amygdala participates in consolidation of the critical item, that item is not susceptible to amnesia. The model assumes that the amygdala plays no role in consolidation of the emotionally neutral items, which is mediated exclusively by the hippocampus.
To accommodate the specific cognitive memory phenomena associated with PTSD, the model requires modification only with respect to effects of the intensity of the emotionally arousing stimulus. That is, the model postulates that the coupling of the amplification of memory for a traumatic stimulus and the decrement in memory for surrounding contextual material reflects the operation of the same neurobiological relationship between the amygdala and hippocampus described above with respect to the von Restorff experiments. If the model is correct, the phenomenology of peritraumatic memory disturbance in PTSD suggests that the amygdala strongly inhibits hippocampal function and that at the highest levels of emotional arousal, the amygdala is the exclusive locus of consolidation of the traumatic event. The failure to consolidate material proximate to the traumatic stimulus (the contextual features of the trauma) due to amygdalar suppression of hippocampal function results in peritraumatic memory impairment.
Traumatic dissociation also is conceptualized in the model as a cognitive phenomenon generated by the proposed interaction of the amygdala and hippocampus. Specifically, dissociation is an extreme form of traumatic memory impairment in which amygdalar inhibition of the hippocampus results in amnesia for the context of the traumatic experience, thereby isolating it from the stream of autobiographical memory. In addition, because consolidation of the central emotional component of the experience occurs in the amygdala alone, memory for the trauma will reflect only those aspects of the experience that the amygdala is able to encode, namely global or gestalt features, rather than the detailed, contextualized information associated with episodic memory mediated by the hippocampus. The result of peritraumatic amnesia and amygdalar consolidation of the trauma might, then, be argued to be the basis of the characteristic phenomenological experience that makes memory for trauma special. Nadel and Jacobs
32 have noted that traumatic memory is stronger than memory for nontraumatic events but is decontextualized from autobiographical memory by means of a complex interaction involving the amygdala and hippocampus. From their point of view, the peritraumatic memory phenomena of PTSD, too, are consistent with the operation of a process in which traumatic experience suppresses hippocampal consolidation and the amygdala becomes the locus of traumatic memory consolidation.
In addition to suggesting a basis for cognitive memory phenomena of PTSD, the proposed model suggests a means to synthesize two conflicting theories of the relationship between the amygdala and hippocampus in emotional memory. One theory argues that the amygdala modulates hippocampal consolidation of memory for emotional stimuli.
33–35 The other theory argues that the amygdala itself is the locus of consolidation of emotional memory.
36 In the new model, contingent on the level of emotional stimulation, the amygdala either enhances or suppresses hippocampal function, and at the highest levels of stimulation, the amygdala increasingly is the primary locus of memory consolidation.
This model avoids the difficulties inherent in any neuroendocrinological explanation for the peritraumatic memory impairment in PTSD. The time frame for occurrence of induced amnesia in PTSD is brief (and in the von Restorff experiments, very brief), suggesting that the relevant mechanisms must be synaptic. Furthermore, the amnestic effect in von Restorff experiments cannot be overcome by simple conscious regulation. Subjects in von Restorff experiments who are educated regarding the amnestic effect and specifically encouraged to attempt to overcome it are unable to do so,
26 suggesting the operation of a reflexive neurological mechanism.
Current understanding of the anatomy and physiology of the amygdala and hippocampus is consistent with the proposed theory. With respect to the hypothesized capacity of the amygdala to consolidate emotional memories, plastic neuronal changes considered necessary features for learning and memory (e.g., long-term potentiation) have been demonstrated in the amygdala.
37 In addition, connections between the amygdala (particularly the basolateral nucleus) and hippocampus that represent potential substrates for the proposed interaction in emotional memory have been demonstrated in rat
38 and monkey.
39Furthermore, established neurophysiological mechanisms are consistent with the proposed biphasic relationship between the amygdala and hippocampus. One candidate mechanism could be analogous to the Renshaw system in the anterior horn of the spinal cord, which, in fact, provided an early model for the electrophysiology of the hippocampus.
40 In this system, low-frequency electrical stimulation of presynaptic neurons elicits postsynaptic action potentials, whereas higher-frequency stimulation results in suppression of postsynaptic activity. The biphasic relationship between frequency and response is explained by the interposition of an inhibitory interneuron between the pre- and postsynaptic neuron. The interneuron functions in a disynaptic feed forward or recurrent collateral feedback circuit activated by presynaptic stimulation. The biphasic response occurs for two reasons. First, the excitation induced by the presynaptic neuron is monosynaptic and therefore precedes the inhibition, which is disynaptic. Second, the duration of the excitatory postsynaptic potential (EPSP) is significantly shorter than that of the inhibitory postsynaptic potential (IPSP). Consequently, at low-frequency stimulation, the sequential EPSPs result in consistent activation of the postsynaptic neuron because stimulation occurs slowly enough to permit postsynaptic recovery from the longer duration IPSPs that follow excitation. As the frequency of presynaptic stimulation increases, the longer duration IPSPs merge additively, overwhelming the shorter duration EPSPs and suppressing activity in the postsynaptic neuron. In fact, it has been demonstrated experimentally, in the cat, for example, that low-frequency electrical stimulation of the amygdala elicits postsynaptic excitation in the hippocampus, which converts to inhibition at higher frequencies of stimulation.
41Various observations in addition to those discussed above with respect to PTSD and memory experiments are consistent with predictions of this model. An independent role for consolidation of emotional memory by the amygdala is suggested by experiments in which neurologically impaired individuals recall emotionally arousing information in the apparent absence of hippocampal involvement.
42,43 Also, cognitive reexperience phenomena, including dreams and waking representations, have been reported to follow emotionally traumatic events for which episodic memory of the trauma has not been consolidated because of closed head or anoxic brain injury.
11,44 These clinical observations of PTSD with neurogenic amnesia are consistent with a theory in which hippocampal consolidation of traumatic stimuli is unnecessary for the development of PTSD.
IMPLICATIONS FOR RESEARCH AND TREATMENT
Laboratory investigations of the relationship between memory intensification and amnesia provide a foundation for a neurobiological model of the cognitive memory phenomena of PTSD. They also suggest a paradigm that would permit the kind of experimental manipulation needed to test hypotheses about the proposed neurophysiological processes. In particular, the von Restorff procedure allows precise variation in the parameters (intensity, duration, emotional valence, sensory modality) of the critical stimulus
29 and is amenable to pharmacological manipulations that are critical in order to elucidate neurophysiological mechanisms. Also, because the von Restorff paradigm is a benign methodology that does not involve fear conditioning, it permits investigation of potential neurobiological mechanisms underlying PTSD in clinical and nonclinical human populations.
Selective pharmacological suppression of the amygdala and hippocampus in nonclinical human subjects could help to establish the role of these structures in memory intensification and amnesia. A reasonable starting point would be to use the von Restorff paradigm to investigate predictions of the model for memory of emotional and neutral material by manipulation of the interaction of the amygdala and hippocampus, as postulated diagrammatically in
Figure 1. Selective pharmacological inactivation of hippocampus or amygdala, in conjunction with functional MRI, could be used to analyze the activity of these structures during exposure to stimuli of varying intensity. Correlational analysis could then test predicted relationships of amygdala activation to the intensification and diminution of memory at various levels of intensity of a critical stimulus.
Proposed treatments for PTSD based on the fear-conditioning model naturally have focused on preventing or extinguishing conditioned fear responses. Treatment approaches aimed at the pharmacological suppression of amygdala activity near to the time of trauma exposure in an attempt to mitigate fear conditioning have been marginally effective.
45 It is our hope that the proposed model, which appears to be more consistent with the true complexity of this often profoundly disabling clinical disorder, may contribute to the development of increasingly effective approaches to treatment.
The proposed model does not represent a comprehensive description of the etiology of PTSD or the reasons that exposure to trauma results in the development of PTSD in some individuals and not in others. Whether or not the model in its current form withstands empirical scrutiny, these and other difficult issues need to be addressed in future work.
One area requiring further study involves investigation of the interactions of the amygdala and hippocampus with regions of prefrontal cortex under conditions of emotional stimulation. There are extensive reciprocal connections between medial and orbital prefrontal cortex and the amygdala, and experimental work has shown that prefrontal cortex can exert inhibitory control over amygdala activity.
46 In addition, specific behavioral effects of prefrontal dysfunction, demonstrated in animals and humans,
47,48 are consistent with the notion that medial prefrontal cortex can regulate amygdala function associated with emotional learning. These findings suggest that the integrity of the prefrontal region may be one factor that determines level of vulnerability for development of PTSD and that the prefrontal cortex may be a potential target for treatment intervention.