The transmissible spongiform encephalopathies (TSEs) are invariably fatal rare neurodegenerative diseases. The most accepted theory is that TSEs are caused by abnormal forms of a naturally occurring glycoprotein. These proteinaceous infectious agents (or prions) are altered forms (PrP
TSE) of the normal cellular prion protein (PrP
C).
3,4 Both isoforms have the same primary sequence, but their three-dimensional conformations are quite different (PrP
C has ∼3% β-sheets, whereas PrP
TSE has ∼43%), as are their biochemical and biophysical properties.
3,5 In particular, the PrP
TSE form is extremely resistant to inactivation by methods ranging from chemical (such as formalin) to heat (including autoclaving) to radiation (both ionizing and ultraviolet).
3 According to the prion theory, infection spreads as a result of PrP
TSE-induced unfolding and refolding of the normal PrP
C into the infectious form. Chaperone proteins may also play a role. There are a number of different PrP
TSE strains with distinctly different clinical and pathological phenotypes. This has been difficult to reconcile with the protein-only prion hypothesis, but may relate to differences in three-dimensional conformation.
3,5,6 The sporadic form of CJD (sCJD), for instance, has at least six major variations based on a combination of different forms of the protease-resistant core of PrP
TSE (type 1 and type 2) and the genotype at codon 129 of the PrP gene (homozygous or heterozygous for methionine or valine).
7,8 These strains vary along many dimensions, including average age at onset, disease duration, constellation of clinical symptoms, neuropathological characteristics, presence of EEG abnormality, diagnostic imaging appearance, and CSF findings.
7,9The TSEs are caused by spontaneous conversion of PrP
C to PrP
TSE (sporadic form), by inherited germ-line mutation (genetic or familial form), and by inoculation (iatrogenic or dietary forms). Sporadic cases (unknown etiology) are the most common (>85%), followed by familial cases (12%–14%); iatrogenic cases are least common.
5 Many different mutations in the PrP gene (
PRNP) have been identified. The predominant forms in humans are Creutzfeldt-Jakob disease (CJD), Gerstmann-Straüssler-Scheinker syndrome (GSS), fatal insomnia (FI), kuru, and new variant Creutzfeldt-Jacob disease (vCJD). Familial GSS is associated with codon 102, 105, 117, 198, and 217 mutations.
10 The familial form of CJD (fCJD) is associated with codon 53, 178, and 200 mutations. The familial form of FI (fFI) is also associated with mutation at codon 178. In addition, the genotype at codon 129 of the PrP gene (homozygous or heterozygous for methionine or valine) drastically affects disease course.
11 Individuals with the codon 178 mutation who are homozygous for valine express fCJD, while those that are heterozygous or homozygous for methionine express fFI.
10,12 Patients heterozygous at codon 129 have a slower disease progression (21±15 months) than those who are homozygous for methionine (12±4 months).
11,1314There is much debate in the medical literature about the existence of some variants of the disease, the true epidemiology, and transmissibility of the infectious agent from one species to another.
15–17 Scrapie has been recognized in sheep from the 1700s onward.
18 A few cases of what would now be considered human TSE were reported in the 1920s and 1930s. The discovery in the 1950s of kuru, with its clinical similarities to scrapie, resulted in a wider recognition of the human forms of TSE. The emergence of bovine spongiform encephalopathy (BSE) in cattle in Great Britain in the 1980s heightened awareness of the TSEs and created concern about the potential for transmission to humans. The identification of vCJD in humans has spurred a resurgence of both concern and research. A PubMed search of TSEs in humans produced 3,636 articles in English; 464 of these were written since January 2001. Currently, many nations have “surveillance units” to follow any suspected cases and to monitor for potential outbreaks.
The worldwide incidence of these rare diseases is estimated as 1 case per million population. Iatrogenic cases have been described in patients receiving cadaver pituitary growth hormone extracts (cadaver extracts are now illegal in most countries, replaced by synthetic growth hormone), insertion of electroencephalographic (EEG) depth electrodes, and corneal and dura mater transplants from persons later found to have infection.
19 Sporadic cases generally occur in the elderly (sixth to seventh decade); the transplant and new variant cases occur in younger adults. Incubation time can be years, with death rapidly occurring once symptoms begin (generally within 9 months of symptom onset).
9,20Although prion diseases are rare, the differential diagnosis includes common conditions such as Alzheimer's disease and vascular dementia. They serve as examples of the importance of newer imaging techniques and diagnostic testing in clinical neuropsychiatry. Additionally, the public health implications of a missed identification of a patient with prion disease are troublesome.
CLINICAL FEATURES
As noted earlier, prion diseases are fatal encephalopathies. The clinical features vary according to the individual diagnosis, and many patients exhibit variations of a central theme according to the form of protein mutation. However, common clinical characteristics exist. An excellent clinical review can be found in the monograph series by Knight and Collins.
21 The following brief overview of this summary can be helpful as an initial framework for study or patient screening in suspected cases.
A diagnostic progression has been developed for CJD in which the diagnosis is definite (confirmed by biopsy or autopsy), probable, or possible. The probables are approximately 95% certain for sporadic CJD, with clinical and EEG or cerebrospinal fluid (CSF) markers. The possibles have less specific clinical and diagnostic findings. The hallmark features of sCJD include a rapidly progressive dementia with visual/cerebellar features (including ataxia), myoclonus, pyramidal/extrapyramidal features, and progression to akinetic mutism. Other vague markers include initial depression, insomnia, or headaches. The variant form has a slower progression, and is reported to have unexplained pain and more psychiatric findings (including delusions, hallucinations, agitation, anxiety, and depression) with less akinetic mutism. Positive EEG and CSF findings are less likely.
21GSS symptoms include a pancerebellar dysfunction with dementia, akinetic mutism, and occasionally aggression or emotional lability. Classic EEG findings are present in some cases. FI has a progressive loss of the normal sleep–wake cycle with eventual inability to attain normal sleep. The patients have hallucinations, parasomnias, and severe sympathetic overactivity (including salivation, rhinorrhea, hyperthermia, tachycardia, and hypertension). Hormonal abnormalities include increased serum cortisol and decreased melatonin or thyrotropin. Abnormal cycles of other hormones including prolactin, and the gonadonal hormones occur. Eventual cerebellar dysfunction, myoclonus, altered respirations, dementia, coma, and death ensue. Polysomnographic testing demonstrates lack of stage 2 and non-REM sleep and only brief REM sleep episodes (often associated with oneiric behavior). Classic EEG and CSF findings are not present. Kuru has a prominent pancerebellar dysfunction with initial ataxia and eventual loss of all voluntary muscle control, gross tremors, movements similar to shivering, euphoria, and late-onset cognitive decline and emotional lability.
21DIAGNOSTIC TESTING
To date, limited medical testing is available to support a proposed diagnosis of TSE.
7 Periodic sharp and slow wave complexes (PSWCs) are frequently found on EEGs but may not be present in the early or late stages of the disease. They are not pathognomonic, as they have been reported in many conditions including Alzheimer's disease. Serial EEGs are recommended in order to have the best chance for demonstration of PSWCs. Test sensitivity is 30% to 100%, and eventual identification of the complexes occurs in approximately two-thirds of sCJD cases. Variant CJD and FI patients do not generally have PSWCs.
21,22Routine CSF studies are normal in prion diseases. The TSEs are unusual in that the immune system is not activated by the infection.
3 Two CSF neuronal proteins (neuron-specific enolase [NSE] and S-100) have been found commonly in patients with prion diseases. However, recent comparisons to another marker, 14-3-3, have established it to be more reliable. 14-3-3 has a sensitivity of 89% to 93% and a specificity of 93% to 100% for sCJD.
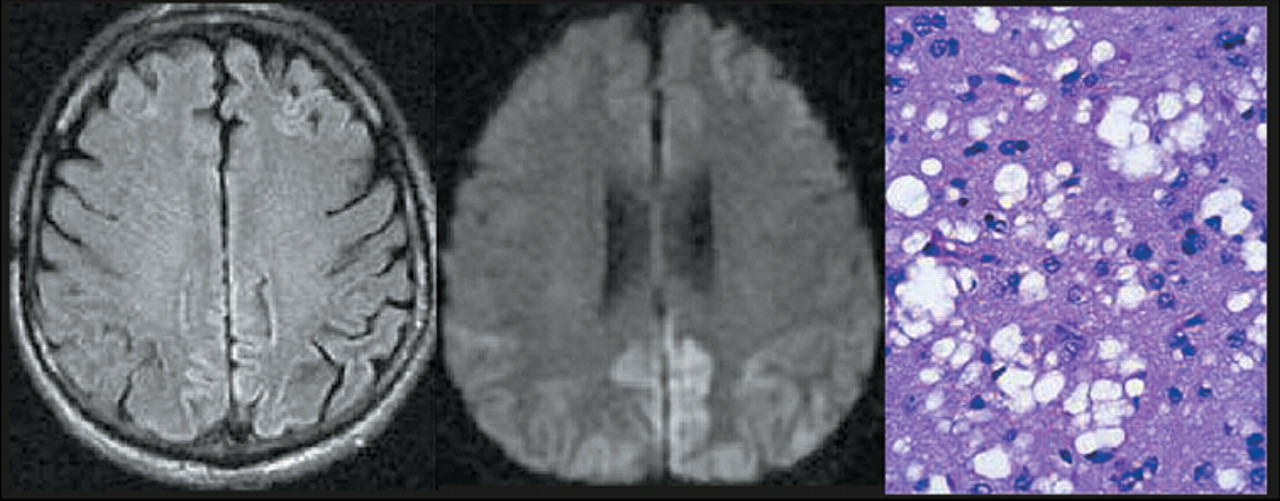
22,23 Several conditions may give false positives, including herpes encephalitis, multiple sclerosis, and cerebral infarction, but they can be eliminated by other diagnostic tests. Although 14-3-3 results are helpful, particularly in cases that do not show all of the classic clinical symptoms, definitive confirmation of the diagnosis requires neuropathological demonstration of the characteristic spongiform changes (
Figure 1).
Computed tomography (CT) is used as screening tool to rule out other illnesses with similar initial presentations. Magnetic resonance (MR) imaging is becoming increasingly useful in the diagnosis of TSEs. A recent large study assessed the presence of high signal intensity within the basal ganglia on T
2-weighted MR imaging in patients with sCJD and those with non-CJD-related dementia.
24 This appearance was found in 67% (109/162) of patients with sCJD but only 7% (4/58) of patients with dementia due to another cause. A similar frequency was found in a previous review of case reports of sCJD with extrapyramidal signs, while an earlier study found increased signal intensity within the basal ganglia in 79% (23/29) of CJD patients.
25,26 Thus the presence of high signal intensity within the basal ganglia has reasonable sensitivity and high specificity for diagnosis of sCJD. High signal intensity may also found in the thalamus (particularly in vCJD) and within the cortical ribbon (
Figure 1). Several studies indicate that a variation on T
2-weighted MR in which the high signal intensity from CSF is removed (fluid attenuated inversion recovery [FLAIR]) is even more sensitive to these changes (
Figure 1).
27 Neuropathological studies indicate that this appearance is associated with gliosis more than spongiform changes.
24,26 Thus T
2-weighted and FLAIR MR imaging would be expected to be most sensitive to TSEs in the later stages of disease progression.
A new type of MR imaging, diffusion-weighted (DW) MR imaging, may provide a way to image changes much earlier in disease progression. Several studies have shown that high signal intensity areas (indicating restricted diffusion) are present on DW MR images prior to the appearance of abnormality on other MR imaging methods (
Figure 1).
28–31 In one study they were present even before the appearance of PSWCs in the EEG or increased protein 14-3-3 in the CSF.
32 Neuropathological studies indicate that this appearance is associated with the microvesicular spongiform changes that occur earlier in disease progression.
31,32 DW MR images are also affected by T
2 weighting, so areas with both restricted diffusion and prolonged T
2 will be even brighter.
28 This may be an additional reason why DW MR images are more sensitive early in disease progression. Sequential imaging in one case demonstrated that very late in disease progression this distinctive appearance on DW MR images can disappear.
30 No histopathology was presented, but the authors suggest that this may be due to the development of gliosis. Thus, DW MR imaging shows promise for both the early diagnosis of TSEs and for monitoring of disease progression.
Functional brain imaging (positron emission tomography [PET], single-photon emission computed tomography [SPECT]) also appears to be quite sensitive to the changes that occur early in the TSEs. Several studies have shown functional decreases (in blood flow or metabolic rate) prior to development of clear abnormalities on structural imaging, EEG, and/or level of 14-3-3 in CSF.
2,33–38 The pattern of abnormalities may be relatively specific to the TSE clinical variant. In sCJD hypoperfusion/hypometabolism primarily in frontal and temporoparietal cortices early in the disease progresses to global decreases by later stages.
33,36,39 Benzodiazepine receptor binding (as measured with
123I-iomazenil SPECT) is also severely decreased, suggesting widespread neuronal degeneration.
39 In iatrogenic CJD (iCJD) cerebellar clinical signs are common.
19 Cerebellar hypoperfusion has been shown early in disease progression.
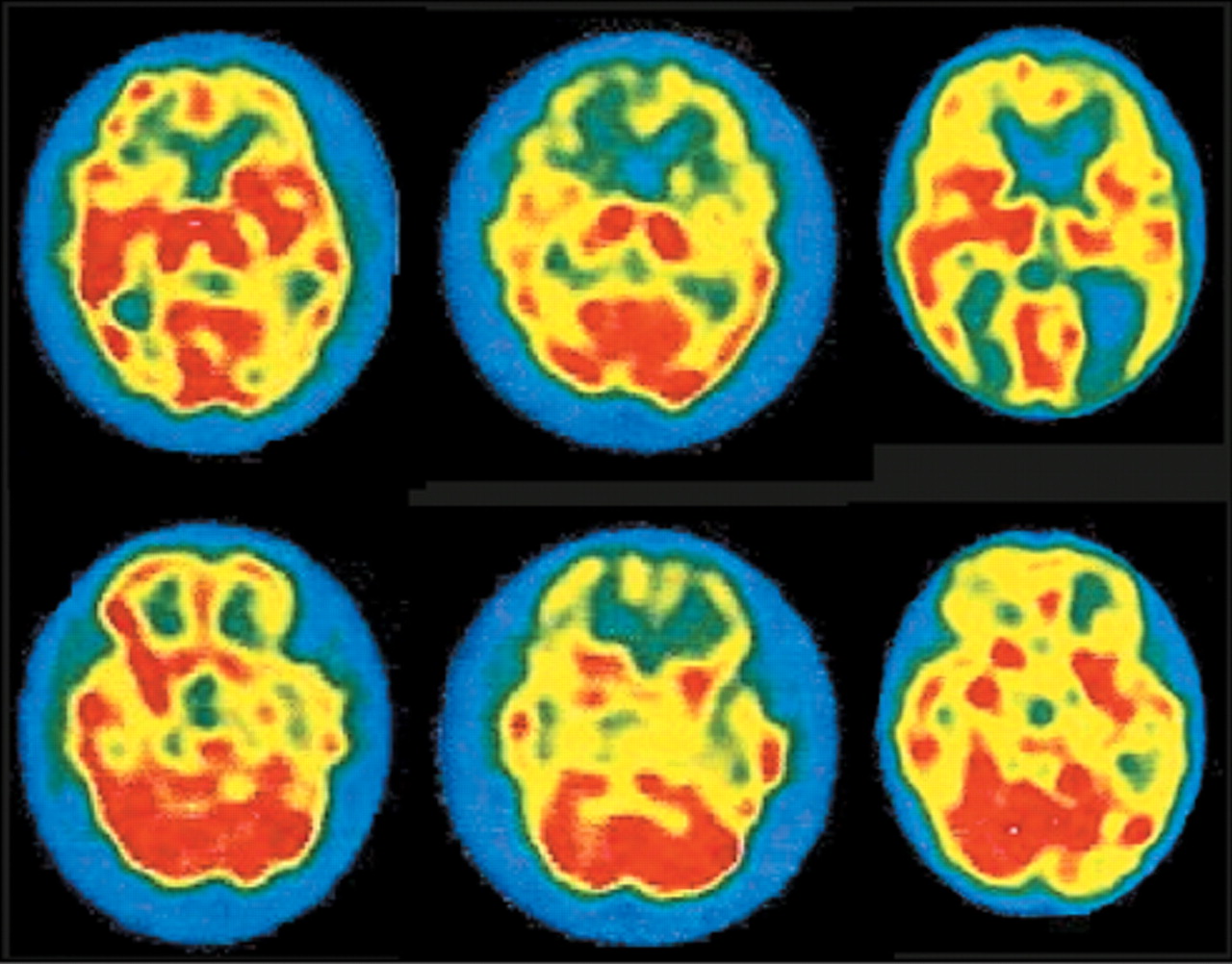
36 In sCJD-Heidenhain variant, which is characterized by visual disturbances (including isolated homonymous hemianopsia), hypometabolism/hypoperfusion is found in occipital cortex (
Figure 2).
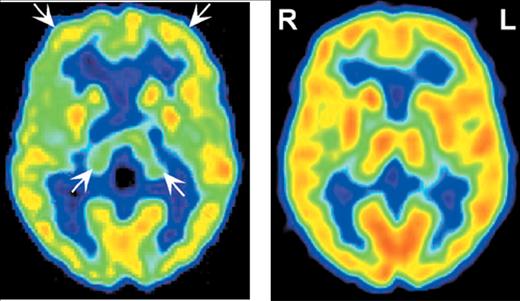
2,37,38 In familial fatal insomnia (fFI) the most common early metabolic changes are in thalamus (particularly anterior) and cingulate cortex (
Figure 3).
11 Neuropathological studies strongly link the autonomic dysfunction and disruption of the sleep–waking cycle that characterize this disease with anterior thalamic degeneration. The authors of this study comment: “The presence of significant hypometabolism in the thalamus and cingulate cortex in all fFI patients, regardless of symptom duration and the codon 129 genotype, indicates that the disease arises relatively early in these structures and subsequently spreads to other areas of the brain.” If this holds true for all the TSE variants, as seems likely, functional imaging may provide a basis for early differential diagnosis.
MR spectroscopy provides another way of assessing cerebral function, by measuring the levels of certain brain metabolites. Most of the few TSE cases examined with this technique were in a fairly advanced state of the disease, with widespread neuronal loss that was reflected by decreases in measured
N-acetylaspartate (NAA, a neuronal marker).
40–42 Two cases have been reported in which NAA was decreased in brain regions that still appeared normal by other techniques.
41,43 In a case of CJD the cortical neuronal number (assessed from a brain biopsy) was normal, but NAA was decreased by 27%.
41 Similarly, NAA was decreased in all areas surveyed (frontal cortex, putamen, cerebellum) in a case of GSS at a time when cerebral blood flow in these areas was normal (as measured by SPECT).
43 Thus MR spectroscopy shows some promise for earlier diagnosis of TSEs and may also help to differentiate TSE from other dementing illnesses.