Niemann-Pick type C disease is an autosomal recessive neurovisceral disorder of lipid storage with a frequency of 1 in 100,000 live births.
4 It is characterized by variable degrees of cognitive decline, behavioral disturbance, and neurological impairment, predominantly ataxia and vertical supranuclear opthalmoplegia.
5 The disease was first described by Niemann in 1914,
6 with its pathology characterized by Pick in 1933.
7 It is biochemically and phenotypically distinct from Niemann-Pick diseases type A and B, which result from a deficiency of lysosomal sphingomyelinase.
8,
9 Genetic analysis reveals two distinct genetic foci, with 95% of disease caused by aberrations in the NPC1 gene on 18q11,
10 coding for the lysosomal NPC1 protein.
11 The less common NPC2 variant is caused by mutations in the NPC2 gene, mapping to chromosome 14q24.3
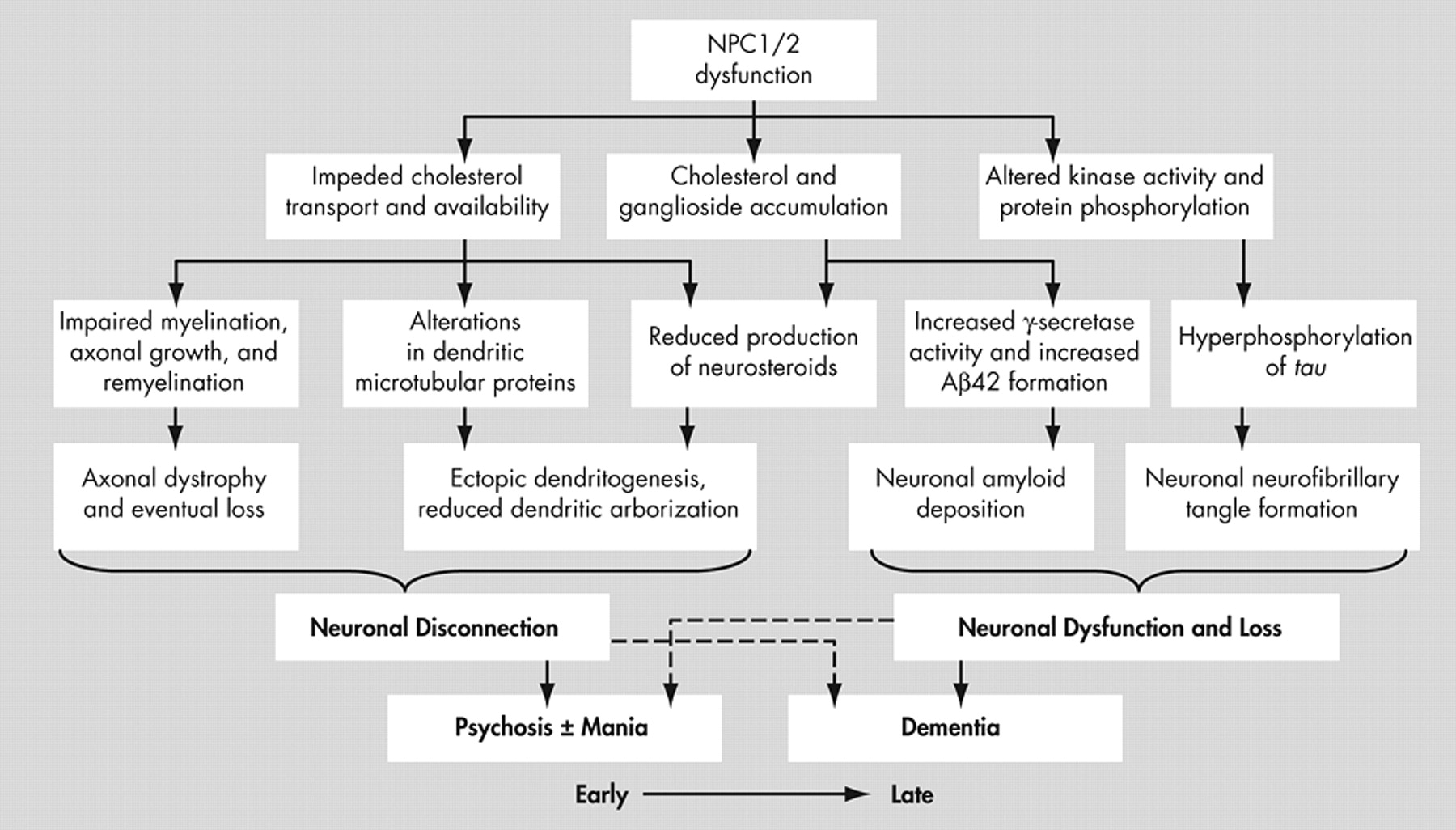
12 and whose product resides in the Golgi apparatus and late endosomes. These proteins are involved in cyclical movement of sterols within cells,
12 –
15 performing cholesterol trafficking and homeostatic functions.
4,
16,
17 Mutation and dysfunction of NPC1 and NPC2 appears to result in late endosomal accumulation of cholesterol, some glycolipids and selected gangliosides
18,
19 leading to Alzheimer’s-like neurofibrillary tangles, neuronal degeneration, neuroaxonal dystrophy and demyelination.
15,
20 –
22 Because of this intracellular cholesterol “traffic jam” and impeded ability to transport endogenously synthesized cholesterol to distal axons, where it is required for membrane maintenance
23 and response to axonal injury,
24 axonal structures are particularly vulnerable and are affected early with axonal spheroid formation, hypomyelination and eventual demyelination.
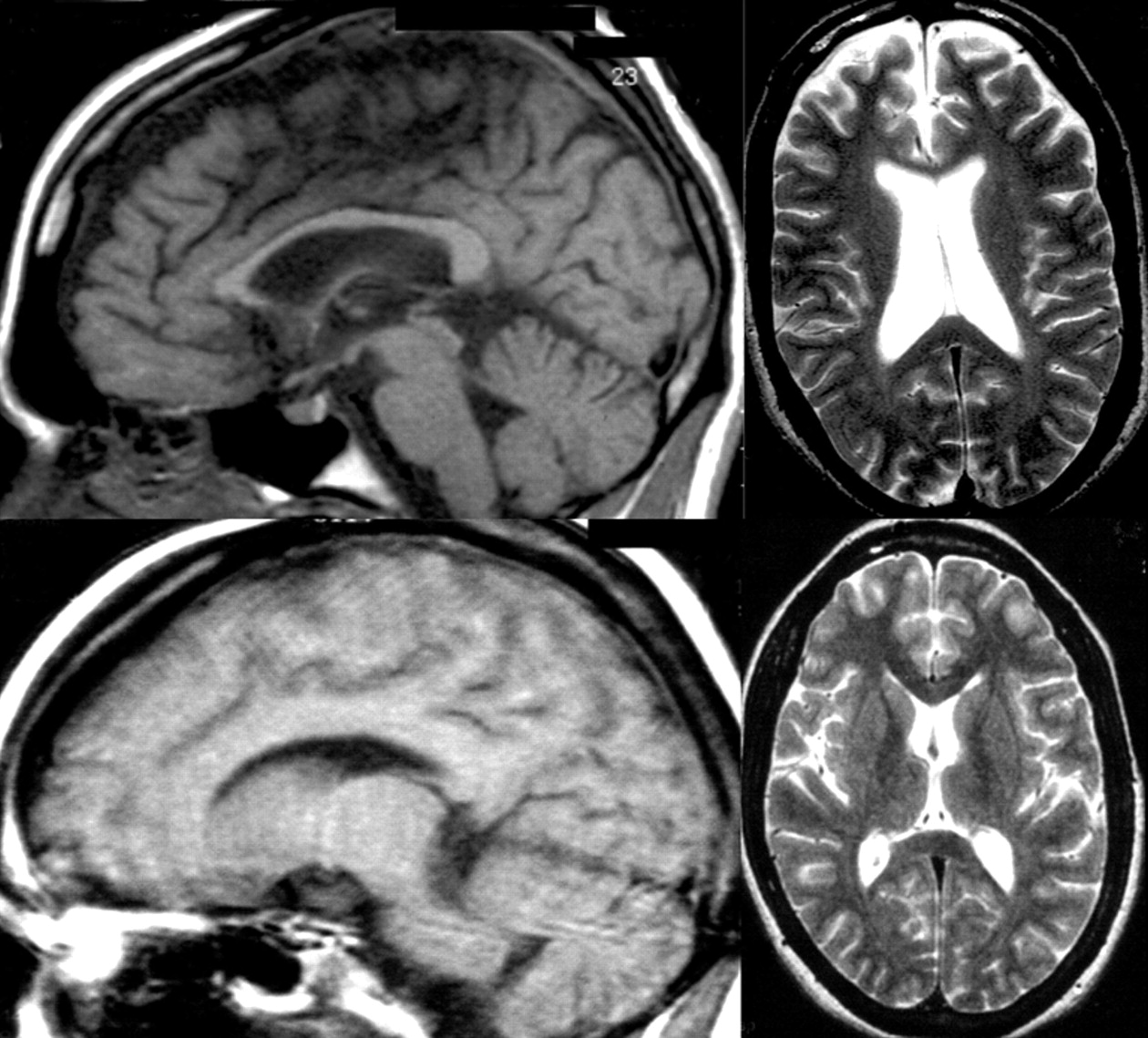
25 As a result, white matter tracts are severely affected,
18,
26,
27 and the corpus callosum may show the most striking axonal loss.
28 Purkinje cells in the cerebellum, basal ganglia, and thalamus are the most vulnerable neurons to Niemann-Pick type C -induced neuronal dysfunction, and these gray matter regions are the first affected, with hippocampal and cortical regions affected later.
26,
29 –
31 Affected neurons, in addition to being swollen and/or affected by neurofibrillary tangle accumulation, often show ectopic dendritogenesis with stunted dendrites and greatly reduced dendritic arborization
32 as a result of altered phosphorylation of the microtubule-associated protein MAP2, which results in dendritic microtubule depolymerization
33 and a reduced availability of arborization-promoting neurosteroids secondary to cholesterol unavailability.
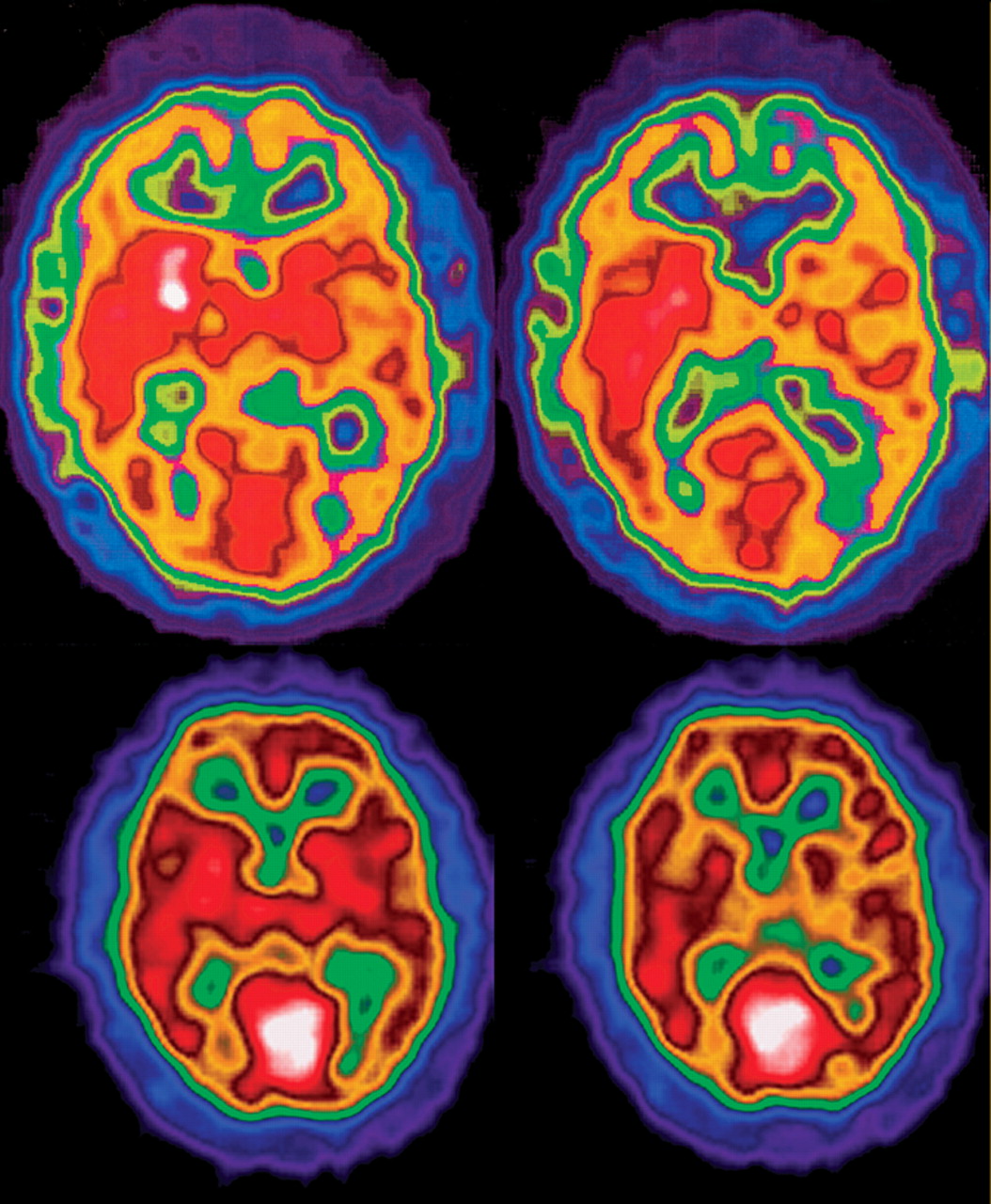
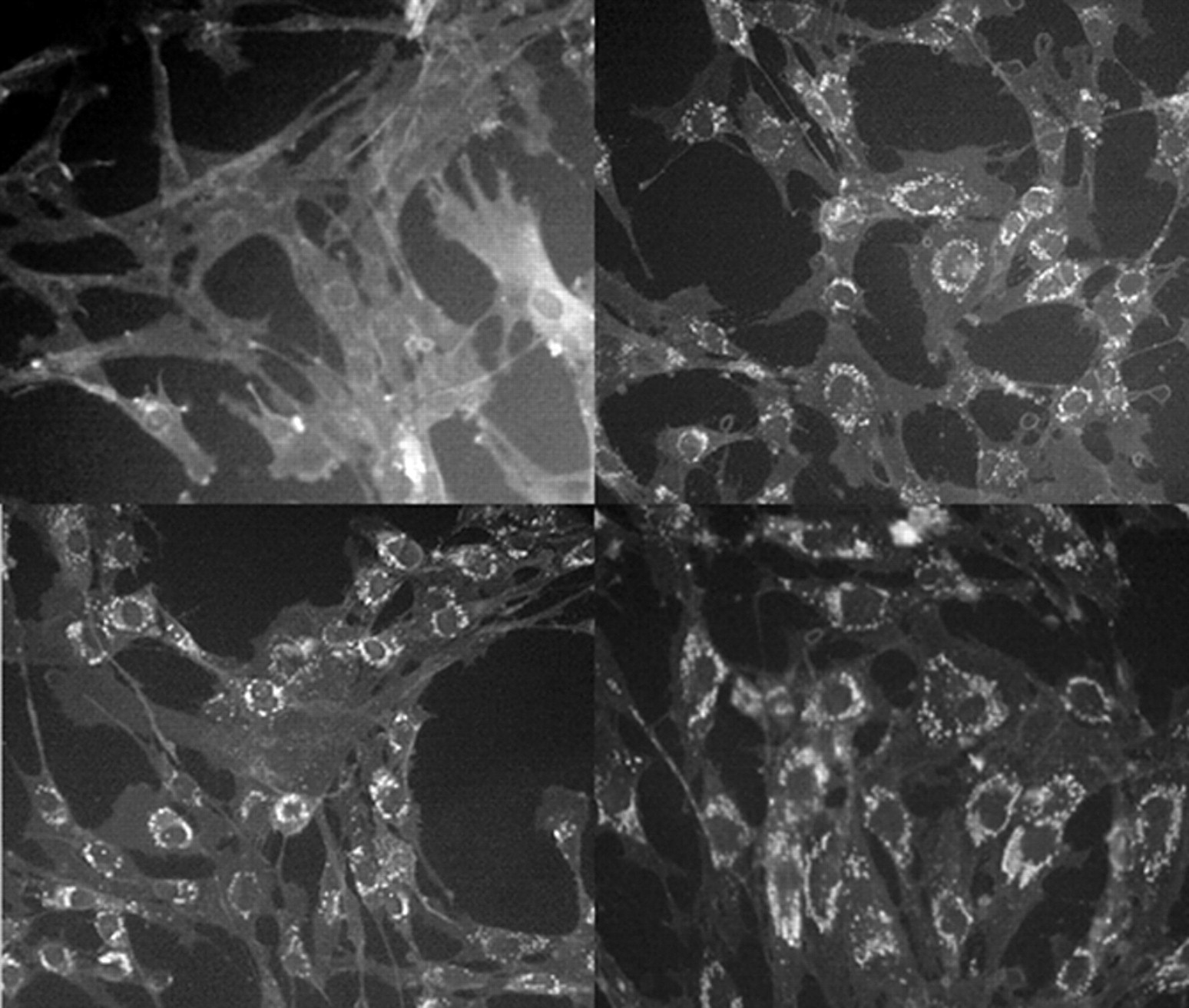
34 The cellular effects of Niemann-Pick type C and their structural and neuropsychiatric sequelae are demonstrated in
Figure 4 .
Niemann-Pick type C may present in infancy, adolescence, or adulthood
37 with a clinically variable picture, although its core features include dementia, dysarthria, ataxia, vertical supranuclear opthalmoplegia and hepatosplenomegaly. It may also commonly present with dystonia and choreoathetosis,
37,
38 although it may occasionally occur without organomegaly.
39 Seizures, dysphagia, and pyramidal signs may appear with disease progression.
37,
38 The range of NPC1 and NPC2 mutations results in marked heterogeneity of clinical presentations.
40 As can be seen by comparison of the clinical phenotype and genotype of cases one and three (
Table 1 ), differing mutations to the same gene may result in disparate neuroradiological, biochemical and psychiatric presentations.