It has been just over two years since the U.S. Food and Drug Administration (FDA) made public the agency's highly controversial analysis of clinical trials data linking antidepressant medications with suicide-related events in children and adolescents. That analysis garnered many supporters as well as numerous detractors and has generated far more questions than it set out to answer.
Last month, physicians, their patients, family members, and advocates got another look at the data and the analytic process that led FDA officials to their conclusions. Tarek Hammad, M.D., Ph.D., who is the senior staff fellow and safety officer in the FDA's Division of Surveillance, Research, and Communications Support that led the agency's 2004 analysis, teamed with Thomas Laughren, M.D., director of the FDA's Division of Psychiatry Products, and Judith Racoosin, M.D., M.P.H., a medical officer and team leader in the agency's Division of Neurology Products, in publishing the analysis as a peer-reviewed report in a major scientific journal.
The article, which appeared in the March Archives of General Psychiatry, detailed the team's review of individual patient-level data from 24 clinical trials on the efficacy and safety of nine antidepressants in 4,582 pediatric patients with major depressive disorder.
While the new report is essentially a condensed version of Hammad's original 131-page analysis of the dataset (Psychiatric News, March 5 and 19, 2004), the tone of the report seems less certain of a direct link between what Hammad and his colleagues call “suicide related events” (SREs) and the medications.
The FDA team analyzed patient-level individual records involving identification of adverse events occurring within the 24 clinical trials that included one or more of a series of terms related to suicidal thoughts, acts, or attempts. Each SRE was then reviewed to determine whether the event described in the record represented an SRE or something else.
A team of suicide experts at Columbia University then categorized all of the SREs into five subclasses of events: events involving a suicide attempt, events indicative of imminent suicidal behavior, events involving suicidal ideation, events involving self-injury with unknown intent, and events involving some sort of injury but was not classifiable as an SRE because of insufficient information. (No suicides occurred in any of the 24 clinical trials.)
The researchers also analyzed a subset of 17 of the 24 clinical trials in which a formal depression rating scale involving a specific suicidality measure was used. They categorized change in suicidality based on any difference between the suicide-rating item at baseline and any point up to the end of the clinical trial. Hammad and colleagues used this dataset to determine “worsening of suicidality” as well as “emergence of suicidality.”
Most of the 24 clinical trials were completed in the latter half of the 1990s and lasted from four to 16 weeks. Sixteen trials involved major depression, four trials involved obsessive-compulsive disorder, two trials involved generalized anxiety disorder, and one trial each involved social anxiety disorder and attention-deficit/hyperactivity disorder.
The analysis identified 109 SREs that occurred within one of the 23 clinical trials conducted by industry and 11 SREs that occurred within the Treatment of Adolescents With Depression Study (TADS), conducted by the National Institute of Mental Health.
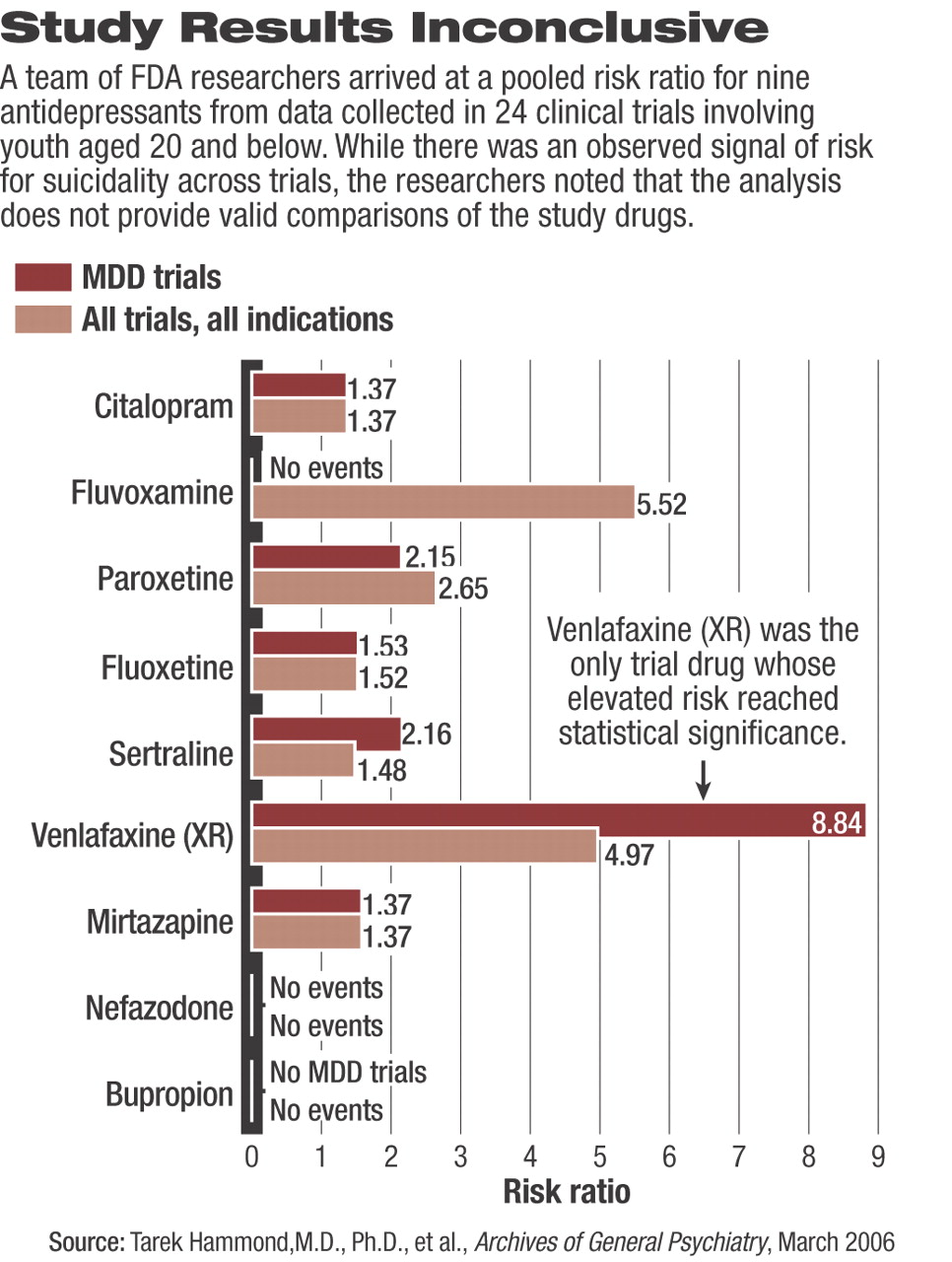
When the researchers analyzed the occurrence of SREs by specific clinical trial, they found SREs occurred in 20 of the 24 trials. Risk ratios for an SRE occurring to a patient taking active medication compared with a patient taking placebo in individual clinical trials varied widely. The lowest risk ratio (RR=0.30—meaning those taking a medication were 70 percent less likely to suffer an SRE compared with those taking placebo) occurred in a trial involving fluoxetine. The highest risk ratio (RR=10.15—meaning those taking medication were 10 times, or 1,015 percent, more likely to suffer an SRE while taking medication compared with those taking placebo) occurred in a clinical trial involving venlafaxine.
Remarkably, however, the only individual clinical trial with a statistically significant increased risk of an SRE associated with the active medication was the TADS trial involving fluoxetine, where patients taking fluoxetine were 4.62 times more likely to have an SRE than patients taking placebo.
With regard to the overall pooled risk of an SRE for each drug (most drugs were investigated in more than one clinical trial), risk ratios ranged from drugs associated with no SREs at all (nefazodone and bupropion) to the highest risk (pooled RR=8.84) associated with venlafaxine (see chart on page 24). Among the nine drugs, venlafaxine was the only one whose elevated risk reached statistical significance.
Of note, the meta-analysis of the 17 clinical trials, which included a standardized measure of change or emergence of suicidality, “revealed no signal for excess suicidality for drug; that is, the [risk ratio] for worsening of suicidality was 0.92 and for emergence of suicidality was 0.93.” Thus, the researchers were troubled that they could not answer the question: If the increased risk is not due to new suicidality and it is not due to a worsening of pre-existing suicidality, then where did the increased risk come from?
The bottom line for the analysis remains roughly the same as it was two years ago. Hammad and his coauthors wrote, “[These results] can be interpreted as indicating that when considering 100 treated patients, we might expect one to three patients to have an increase in suicidality beyond the risk that occurs with depression itself, owing to short-term treatment with an antidepressant.”
Hammad, Laughren, and Racoosin were careful to emphasize the limitations of their analysis, most significantly the post-hoc nature of the analysis in which multiple outcomes and many subanalyses tend to increase the level of uncertainty of the results. They also cautioned readers about overgeneralizing the results in these 24 short-term clinical trials to patients in the general public, who are significantly more likely to be taking these medications for significantly longer periods of time.
Additionally, Hammad and his coauthors emphasized that the analysis“ cannot provide valid comparisons of the nine drugs studied.”
They noted that the data were pooled to increase the statistical power of the analysis for determining the suicidality risk of the nine antidepressants as a whole or taken together.
Despite these and a few other noted minor limitations, the FDA team concluded that “the observed signal of risk for suicidality represents a consistent finding across trials, with many showing RRs of two or more.” Nonetheless, they cautioned, “an overall interpretation of this finding and its implications for clinical practice are less clear.”
Arch Gen Psychiatry 2005 63 332
