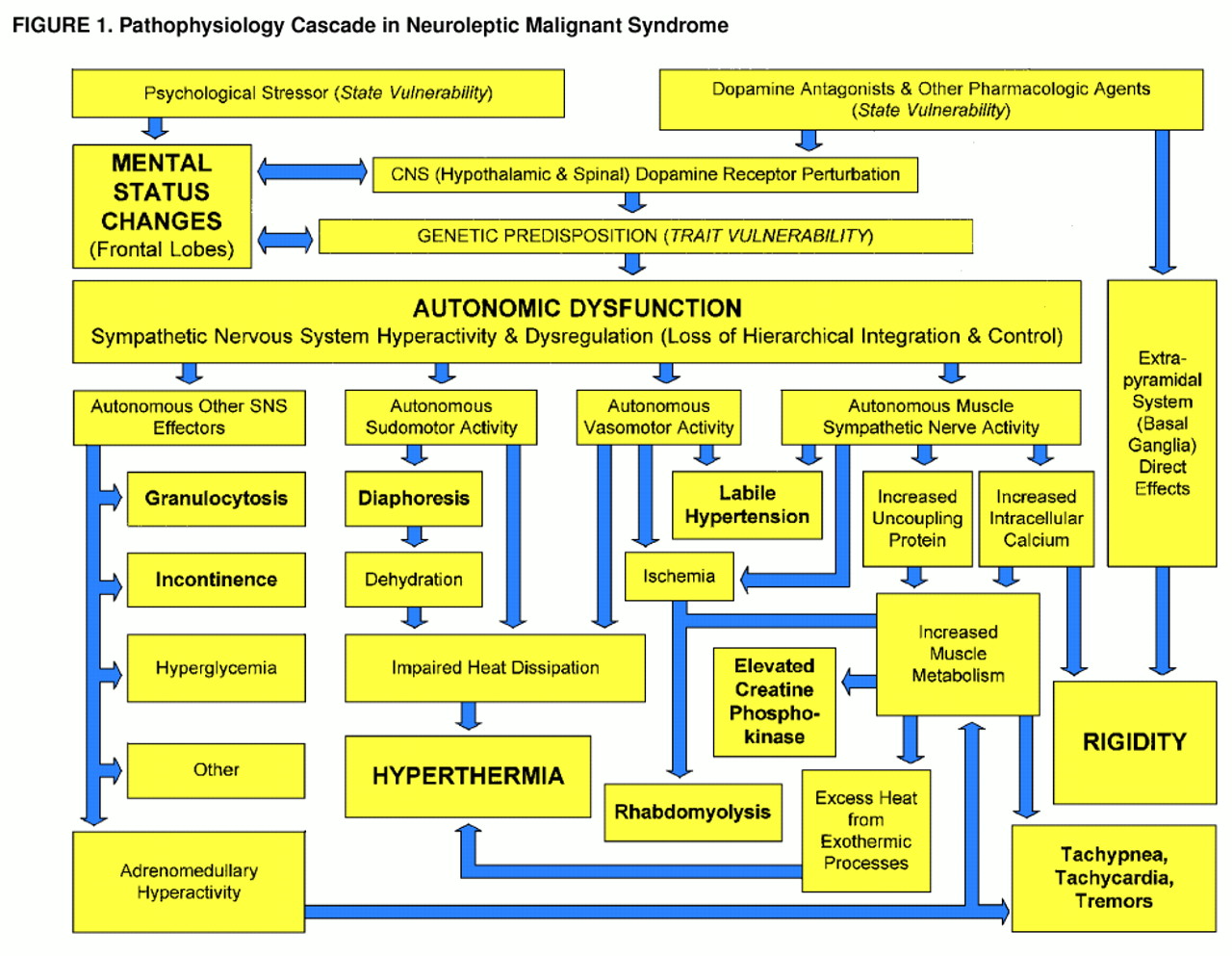
Altered mental status
Altered consciousness is considered by some
(120) to be a sine qua non for the diagnosis of neuroleptic malignant syndrome. In one relatively large series
(98), “a striking, frightened facial expression was observed in all cases,” accompanied by “a sense of doom” and “overwhelming anxiety” (p. 719). Tollefson
(6) described a mute and akinetic patient with neuroleptic malignant syndrome as having “an exaggerated startle response.” Catatonic patients retrospectively report intense, uncontrollable anxieties
(104). Healthy volunteers viewing a frightening film have increased plasma norepinephrine, but their other catecholamine and endocrine levels are normal
(121). Psychological stress is positively correlated with plasma vanillylmandelic acid but not 3-methoxy-4-hydroxyphenylglycol, homovanillic acid, or 5-hydroxyindoleacetic acid, suggesting that the sympathetic nervous system is activated more by transient emotional stress than by persistent emotional conditions
(122).
Acute, but not subacute or chronic, psychosis and Brief Psychiatric Rating Scale scores indicating global psychopathology and anxiety are positively correlated with serum creatine phosphokinase levels
(123). Catatonic excitement has been promoted as a highly reliable risk factor for neuroleptic malignant syndrome
(102), and labile mood and insomnia consistently precede lethal catatonia
(124). “Emotional disturbance” can trigger hyperthermia and many of the clinical features usually associated with neuroleptic malignant syndrome
(125), as can acute phencyclidine-induced psychosis
(126). Intense emotional or psychological disturbance, with altered frontal cortical function, could be a pathophysiological link between these syndromes and neuroleptic malignant syndrome.
Dysautonomia
Diaphoresis is common in neuroleptic malignant syndrome
(124), with rates from 50% to 100%
(98,
99,
105), and is due to direct sympathetic nervous system stimulation rather than circulating catecholamines
(17,
36). In contrast to its role in true fever, diaphoresis in neuroleptic malignant syndrome is not part of a coordinated effort to lower body temperature
(127). Eccrine sweat glands are the only mammalian organs with a purely thermoregulatory function
(23) and have the capacity to dissipate heat faster than it can be generated
(128), so hyperthermia in neuroleptic malignant syndrome implicates a defective heat-loss mechanism in addition to any increase in thermogenesis. Excessive sweat gland activity is probably responsible for neuroleptic-malignant-syndrome-associated dehydration in some cases
(129), and dehydration may contribute to hyperthermia.
Tachypnea and tachycardia in neuroleptic malignant syndrome reflect a hyperadrenergic state, but increased metabolism makes additional demands on the cardiopulmonary system. In one drug-induced syndrome similar to neuroleptic malignant syndrome, body temperature increased in tandem with blood pressure and heart rate
(130), and body temperature accounts for as much as 16% of the respiratory rate variance in neuroleptic malignant syndrome
(127).
Urinary incontinence is another clinical manifestation of autonomic dysfunction in neuroleptic malignant syndrome. The bladder base and internal sphincter consist of smooth muscle innervated exclusively by predominantly α-adrenergic sympathetic nervous system fibers. Voluntary restraint of micturition requires the frontal lobes, and lesions in either hemisphere can cause bladder incontinence
(17,
67). In spinal cord patients, diaphoresis and bladder incontinence are associated with flexor spasms of the lower extremities, suggesting an intrinsic relationship among these motor responses
(27).
Catecholamine excess alone can produce a syndrome similar to neuroleptic malignant syndrome, as illustrated by the case of a 34-year-old woman who experienced the sudden onset of palpitations, inability to move or speak, and labile postural tachycardia and hypertension
(111). Mutism, increased muscle tone, hyperactive tendon reflexes, and catalepsy were reproduced reliably by physical activity and anxiety-provoking situations and were always associated with hypertension and tachycardia. Plasma epinephrine and norepinephrine were markedly elevated and correlated with amount of time upright, but urine 24-hour catecholamine and monoamine metabolite levels were normal. She was found to have a hyperactive sympathetic response and peripheral α
1-adrenoceptor subsensitivity
(111).
Hyperthermia
Hyperthermia can occur when the heat-loss apparatus is defective or when intact thermoeffectors are poorly coordinated. The ineffectiveness of diaphoresis, normally a potent cold effector, in neuroleptic malignant syndrome has already been discussed. The simultaneous activation of both warm and cold effectors in neuroleptic malignant syndrome indicates that although thermoeffectors remain operational, their activities are not coordinated
(76,
127,
131). This condition alone would be sufficient for hyperthermia to supervene, but in addition there are two likely sources of excess thermogenesis in neuroleptic malignant syndrome: accelerated brown-adipose-tissue-like metabolism (uncoupled phosphorylation) and excessive catecholamine-induced Ca
2+ release from the sarcoplasmic reticulum.
Persistent stimulation of sarcoplasmic reticulum Ca
2+ release would lead to continuous activation of Ca
2+ reuptake mechanisms, releasing heat as a by-product. This could account for the observation that serum creatine phosphokinase levels are positively correlated with 24-hour urinary catecholamine metabolite excretion in neuroleptic malignant syndrome
(16). The appearance of elevated muscle enzyme levels in neuroleptic malignant syndrome before clinical signs of autonomic instability are evident indicates that hypermetabolism may precede the full syndrome
(132). Hypermetabolism related to hyperthyroidism, in the absence of central dopamine blockade, can produce a syndrome similar to neuroleptic malignant syndrome
(133).
Peripheral catecholamines also directly stimulate uncoupled phosphorylation in mitochondria, as occurs in some cases of pheochromocytoma, cold exposure, and treatment with sympathomimetic agents. As in these other examples, prolonged elevation of peripheral catecholamine levels in neuroleptic malignant syndrome may even lead to induction of uncoupling protein mRNA and increased thermogenic tissue mass, suggesting a possible physiological basis for persistent hyperthermia during recovery from neuroleptic malignant syndrome. This thermogenic mechanism is distinct from that related to intracellular Ca
2+ homeostasis. Hyperthermia in neuroleptic malignant syndrome can be dissociated from muscle rigidity
(33,
134) and creatine phosphokinase levels
(135), precede extrapyramidal signs
(115), and persist despite curarization-induced flaccidity
(33). The lack of glycogen and lipid stores in muscle biopsies strongly suggests that uncoupled phosphorylation contributes to hyperthermia in neuroleptic malignant syndrome
(136).
Rigidity, elevated creatine phosphokinase, and rhabdomyolysis
Asymptomatic creatine phosphokinase elevations occur in patients with psychotic disorders, whether clinically stable or acutely ill, following oral treatment with typical or atypical neuroleptics
(137–
139). Clozapine is associated with more frequent and higher creatine phosphokinase elevations than are typical neuroleptics
(139); this is noteworthy because clozapine has significantly greater α
2-adrenoceptor activity
(140), which is probably the cause of de novo diabetes mellitus in clozapine-treated patients
(141). Substantial creatine phosphokinase elevations accompanied only by tachycardia and elevated blood pressure may follow neuroleptic treatment
(142), suggesting a hypermetabolic effect mediated by the sympathetic nervous system.
Cardiac lesions caused by elevated catecholamines are indistinguishable from atherosclerotic lesions
(143). Short-term hypoxia permits creatine phosphokinase-MB to escape without necrosis
(144), and intravenous isoproterenol causes elevated creatine phosphokinase-MB in childhood asthmatics
(145). Elevated creatine phosphokinase and focal skeletal myositis occur in pheochromocytoma
(146), which releases mainly norepinephrine
(119). Ischemia mediated by catecholamine-induced vasoconstriction in skeletal muscle is probably the cause of muscle injury in pheochromocytoma
(147). Creatine phosphokinase elevations and myoglobinuria in neuroleptic malignant syndrome may exist without rigidity or tremor
(148), and haloperidol-induced rhabdomyolysis is associated with autonomic dysfunction but not rigidity
(149).
Urinary catecholamines and catecholamine metabolites are positively correlated with blood creatine phosphokinase levels in acute neuroleptic malignant syndrome, but other clinical features are not
(16). In patients with neuroleptic malignant syndrome, serum creatine phosphokinase covaries nonlinearly with other muscle enzymes and exceeds by as much as 15-fold the normal extracellular-intracellular gradient, indicating that muscle necrosis alone cannot account for elevated creatine phosphokinase levels
(150). Experimental tissue injury models suggest that increased enzyme synthesis represents a reparative response to a noxious agent
(151). The noxious event in neuroleptic malignant syndrome could be a relative hypoxia created by a metabolism-perfusion mismatch caused by a hyperactive and unregulated sympathetic nervous system.
Catecholamine-induced Ca
2+ release from the sarcoplasmic reticulum may also cause cell death
(152). Focal myocardial necrosis occurs in patients with pheochromocytoma
(153), in rats infused with epinephrine and norepinephrine
(143), and in dogs infused with inotropic amines
(154). Muscle necrosis in these settings is apparently due to a direct effect of catecholamines on Ca
2+ influx
(81,
153). Oxidation of excess catecholamines to catecholamine-O-quinones, with subsequent formation of superoxide radicals that attack cellular membrane and protein constituents essential to energy metabolism, is another mechanism by which excess catecholamines can damage myocytes
(155).
Muscle biopsy findings in patients with neuroleptic malignant syndrome have yielded minimal or inconsistent results (compare references
6 and
156 with references
34,
120,
136, and
157). Muscle abnormalities that have been detected in neuroleptic malignant syndrome resemble those found in malignant hyperthermia
(157) or consist of endomysial edema with a striking absence of both glycogen and lipid substrate stores
(136). Profound depletion of intracellular energy stores strongly implicates uncoupled phosphorylation as the source of hyperpyrexia
(136), and the resemblance to malignant hyperthermia in these cases implicates Ca
2+ overload as a common etiological factor. A majority of patients with severe neuroleptic malignant syndrome have documented serum hypocalcemia
(98), which could reflect a sudden net influx of Ca
2+ into myocytes that coincides with a “malignant” phase of the syndrome.
Extrapyramidal signs associated with neuroleptic malignant syndrome include rigidity, tremor, cogwheeling, dystonia, chorea, dyskinesia, opisthotonos, opsoclonus, and posturing
(3,
5, DSM-IV). As the data presented here indicate, rigidity in neuroleptic malignant syndrome could be caused by catecholamine-induced changes in intracellular [Ca
2+], and tremors could also reflect elevated peripheral catecholamines. Increased muscle tone caused by sympathetic nervous system hyperactivity is independent of, but may coexist with, effects of neuroleptics on the extrapyramidal system, and the presence of more complex motor signs implicates concomitant basal ganglia involvement in some cases of neuroleptic malignant syndrome.