Risperidone has a cumulative worldwide use exceeding one million man years. It is common clinical practice to use risperidone before or instead of a trial of clozapine in treatment-unresponsive patients. There are, though, scant data to support the superiority of risperidone in populations that do not respond to conventional compounds
(1–
16).
Clozapine is a definite advance in the treatment-refractory schizophrenic individual, but its high cost, myelotoxicity, requirement for continuous granulocyte monitoring, and myriad other side effects have greatly limited its use among both clinicians and patients. Since its U.S. approval for general use in late 1989, clozapine has been used in slightly more than 140,000 patients. Considering that about 800,000 schizophrenic patients in this country are intolerant of or unresponsive to conventional antipsychotics, clozapine is clearly underused and is likely to remain so into the foreseeable future. Indeed, since the approval of risperidone in late 1993, nearly one-third of the patients who were taking clozapine have been changed over to risperidone.
In the pivotal, industry-sponsored trial before Food and Drug Administration approval, risperidone’s efficacy was shown in patients with known historical response to conventional antipsychotics (13). Close examination of the length of stay of patients recruited for this trial revealed that a significant proportion (N=93 of 523) of these individuals had been hospitalized continuously for more than 6 months (average=104 weeks) before study entry. Curiously, these long-hospitalized subjects had a robust response to only the 6-mg/day dose of risperidone. The other doses of risperidone (2 mg/day, 10 mg/day, and 16 mg/day) and haloperidol (20 mg/day) were ineffective in this subgroup of 93 chronically hospitalized patients. These data suggested that risperidone may be more effective than haloperidol in treatment-refractory subjects. They further hinted that the dose response curves for risperidone were distinct for populations that were responsive and refractory (to conventional medications).
Bondolfi and colleagues
(3) compared clozapine to risperidone in 86 “relatively” refractory schizophrenic subjects by using a randomized, double-blind, parallel group design. The results revealed a high response rate for both groups (approximately 65% of subjects had greater than 20% improvement from baseline), but neither drug was superior in treating either positive or negative symptoms. Risperidone appeared to work faster than clozapine but probably only because risperidone can be titrated to efficacious levels more rapidly than clozapine. The goal of our study was to compare the relative safety and efficacy of risperidone and haloperidol in a population of carefully defined subjects with treatment-refractory schizophrenia.
METHOD
Subjects
Sixty-seven subjects were recruited from two facilities, the West Los Angeles Veterans Administration (VA) Medical Center and Camarillo State Hospital. After complete description of the study to the subjects, they signed informed consent documents that were approved at their particular institutions. In situations in which subjects had conservators, consent was obtained from both the patient and the conservator. Subjects were included only if they were 18–60 years of age, had a diagnosis of schizophrenia, were able to take oral medication, and, in the opinion of the investigator, were able to adhere to the required schedule of evaluations. The primary diagnosis was established by employing DSM-III-R criteria as determined from a review of the chart history and by using symptoms elicited during the Structured Clinical Interview for DSM-III-R—Patient Version (SCID-P)
(17). SCID-P and Brief Psychiatric Rating Scale (BPRS) interviewers were trained at the Diagnosis and Psychopathology Unit of the University of California at Los Angeles Clinical Research Center for the Study of Schizophrenia and participated only when ratings met the center’s minimum criteria for reliability (kappa ≥0.75 for SCID-P; intraclass correlation coefficient ≥0.80 for BPRS).
The minimum symptom severity for study entry was identical to that proposed by Kane et al.
(18): a total BPRS
(19) score of at least 45 (18-item version; 1=absent, 7=severe)
(20) and a minimum Clinical Global Impression (CGI)
(21) scale rating of 4 (moderately ill). In addition, item scores of at least 4 (moderate) were required on two of the four “psychotic” items from the BPRS (conceptual disorganization, suspiciousness, hallucinatory behavior, and unusual thought content). All subjects met treatment-refractory criteria, which entailed a failure to respond to or an inability to tolerate at least three 6-week epochs of treatment within the preceding 5 years with antipsychotic medications from at least two different chemical classes, at daily doses equivalent to or greater than 1000 mg of chlorpromazine. These were the same criteria used by Kane et al.
(18) in their study that established the efficacy of clozapine in treatment-refractory individuals.
Subjects were excluded from the study if they had significant medical disease, had a history of seizure disorder, or had taken any investigational drug during the 4 weeks before the start of the study. Subjects could not have physical or cognitive impairment of such severity as to adversely affect the validity of clinical ratings or impair capacity to give informed consent. Subjects with substantial substance abuse during the 2 months before the study or with substance dependence 6 months before the study were excluded. Finally, subjects with a high risk of violence directed toward themselves or others or a history of risperidone treatment failure (due to either nonresponse or intolerance) were also excluded from entry.
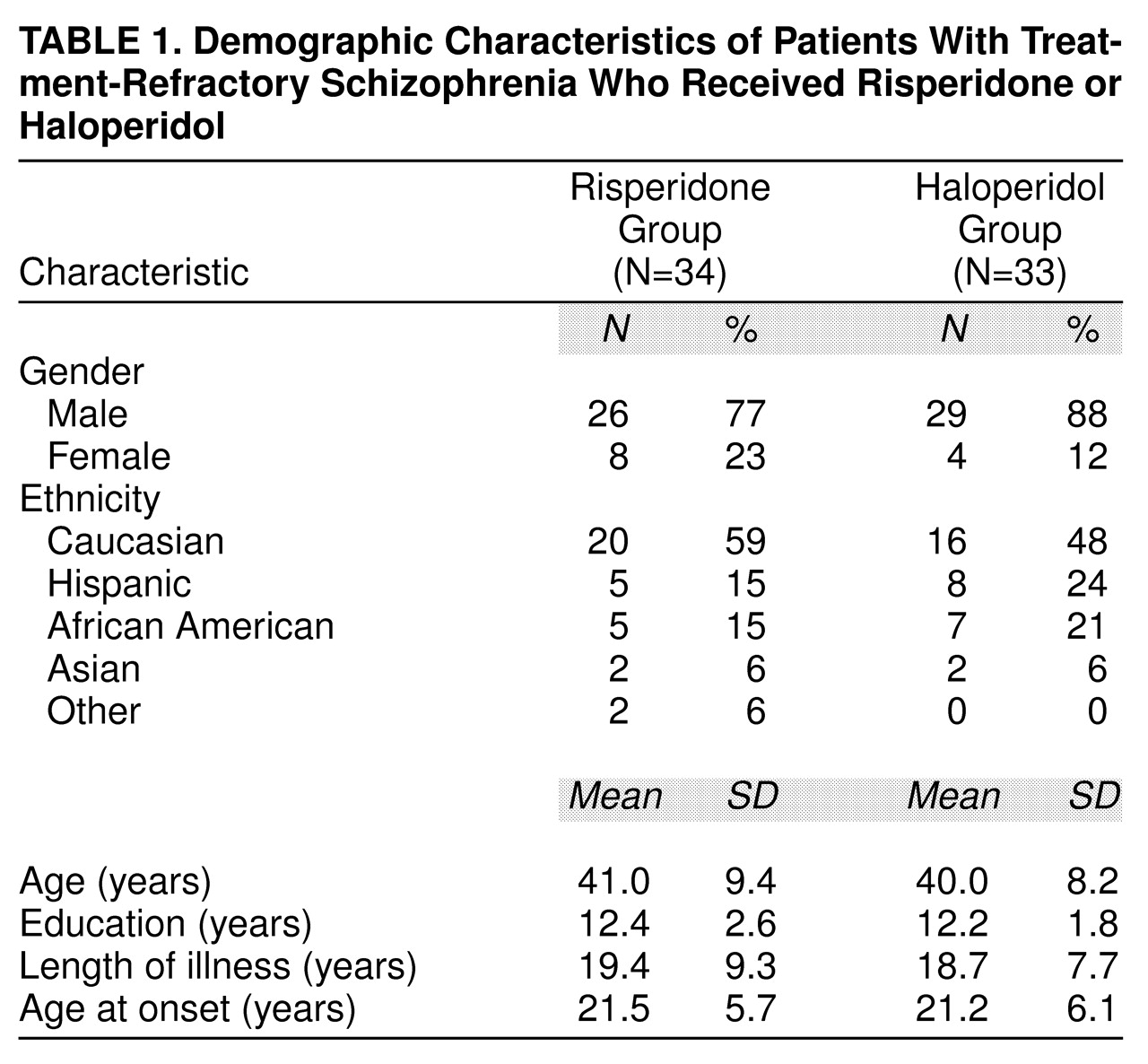
The demographic characteristics of the study population are listed in
table 1. There was no statistically significant difference between the risperidone- and haloperidol-treated patients on demographic factors.
Procedure
All patients completed a 3–7-day, single-blind, placebo washout phase. No subject met criteria for responsiveness to placebo (greater than 20% improvement from baseline). Following this phase, subjects were randomly assigned to double-blind treatment with either risperidone, 6 mg/day, or haloperidol, 15 mg/day, for a 4-week fixed-dose phase. Random assignment to drug groups was performed through use of a computerized random-number-generating program. We chose 6 mg of risperidone because greater response was seen at 6 mg in the chronically hospitalized subgroup treated in the pivotal phase III study
(13). We chose 15 mg of haloperidol because in our group’s experience, it provided the most reasonable compromise between efficacy and toxicity
(19). Although the 20-mg/day dose of haloperidol does show slightly overall better efficacy than the 10-mg/day dose, it is associated with substantially more treatment intolerance and observable extrapyramidal toxicity. Pharmacotherapy during baseline measures differed between the two sites. Baseline assessments at the state hospital site were conducted while patients received 15–30 mg/day of haloperidol at the end of the 3-week haloperidol stabilization phase. Baseline assessments at the VA hospital were conducted during the placebo lead-in period. This difference potentially could have increased the variability in the pooled cohort at baseline but would have had little influence on any differential treatment effects, since patients were randomly assigned to treatment within each site. No baseline differences between sites were detected on psychopathology and side effect ratings.
The number of pills was constant for each group throughout both the fixed and flexible phases. The fixed-dose phase was followed by a 4-week, flexible-dose phase in which clinicians could choose to blindly increase or decrease the doses of risperidone between the ranges of 3–15 mg/day or haloperidol between the ranges of 5–30 mg/day depending on the patients’ clinical condition. After ratings were complete for this flexible-dose phase, the blind was broken, and patients assigned to haloperidol were given an opportunity to try risperidone.
Subjects could receive anticholinergic medications (either benztropine or biperiden) for parkinsonism or dystonia or propranolol for akathisia. For severe, acute anxiety or agitation, subjects could receive lorazepam on an as-necessary basis, up to 8 mg/day. Temazepam could be prescribed for insomnia.
Psychopathology and extrapyramidal side effects were assessed at baseline and once every 2 weeks thereafter. Measures included the 24-item BPRS
(20,
21) and the CGI
(22). The latter instrument contained both physician-rated and patient-rated components. Extrapyramidal side effects were assessed with the Barnes Akathisia Scale
(23), the Abnormal Involuntary Movement Scale (AIMS)
(24), and the Simpson-Angus Scale
(25).
Statistical Analyses
The statistical comparison of drugs was performed by using analyses of covariance. Analyses were done separately in the fixed- and flexible-dose phases, with last observation carried forward. Analyses were performed only on subjects who completed at least 1 week in a particular phase. The dependent variables were change from baseline on several measures of psychopathology and extrapyramidal side effect variables; baseline measures were used as covariates. A second set of analyses examined the percentage improvement from baseline. Chi-square analyses were used to determine if there were differences between drug treatment groups (risperidone versus haloperidol) in the use of concomitant medication. To determine correlates of response to risperidone, chi-square analyses and t tests were performed to compare, on a number of baseline characteristics, those patients who demonstrated more than a 40% improvement in BPRS scores at the end of the flexible phase with patients who did not.
RESULTS
The dropout rates for both drug groups were small, and there was no statistically significant difference in the overall dropout rates (six risperidone-treated versus five haloperidol-treated individuals). All five of the haloperidol-treated subjects were dropped because of lack of drug efficacy, whereas subjects from the risperidone group were divided between those who dropped out because of lack of drug efficacy (N=3) and because of treatment-emergent side effects (N=3). Two of the risperidone-treated subjects dropped out because of extrapyramidal side effects, and one dropped out because of erectile incompetence. The average doses of risperidone and haloperidol were 7.5 mg (SD=1.9) and 19.4 mg (SD=5.6), respectively, during the flexible-dose phase.
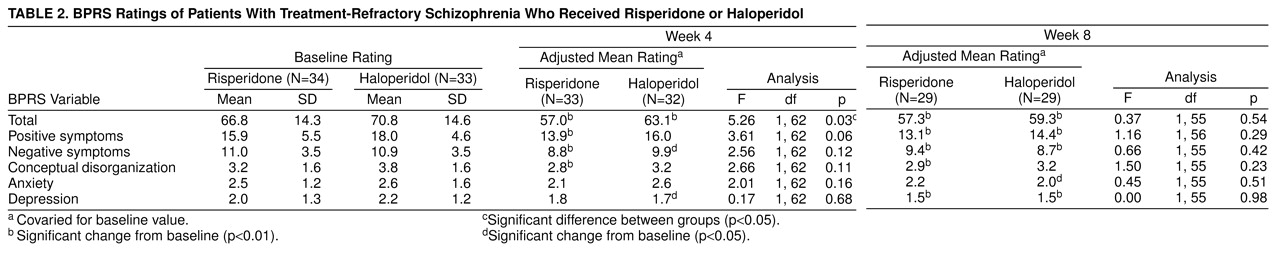
There was a statistically significant difference in improvement between baseline and the end of the fixed-dose phase on overall psychopathology as measured by the total BPRS score (p=0.03), favoring risperidone (
table 2). No advantage was seen, however, at the end of the flexible-dose phase for risperidone-treated subjects. Overall, at the end of the flexible phase, risperidone-treated patients improved 24% (as measured by reduction of total BPRS scores), whereas haloperidol-treated subjects had a 10% improvement (F=5.61, df=1, 62, p=0.02).
The group distribution of improvement on the BPRS at the end of the fixed-dose phase appeared bimodal, suggesting that the risperidone-treated patients may have been drawn from two distinct populations. The larger subgroup (70%, N=23, of the study group) had a mean improvement of 10% on the BPRS, whereas the remainder had a robust clinical response with a mean improvement of 55% over baseline. Unfortunately, perhaps because of the small group size, a two-population mixture model with nonlinear regression could not confirm this observation statistically.
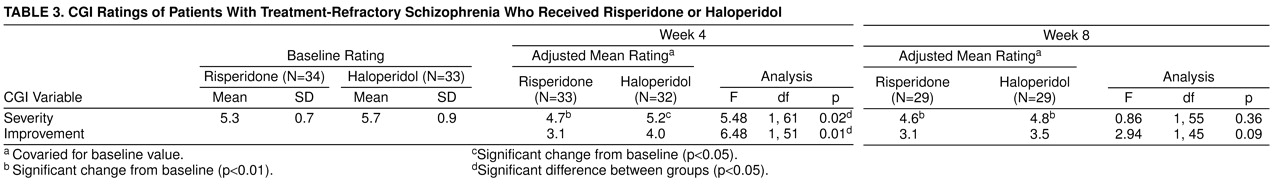
At the end of the fixed-dose phase, 13 (42%) of 31 risperidone patients were judged by their clinicians as having demonstrated mild or greater improvement with few or no side effects, compared with six (17%) of 32 haloperidol patients (χ
2=4.02, df=1, p=0.05). At the end of the flexible phase, the comparable figures were 28% (N=7) among risperidone patients and 25% (N=7) among haloperidol patients; the difference was not significant (
table 3). CGI ratings by the patients themselves revealed a somewhat more positive picture for risperidone. At the end of the 4-week, fixed-dose phase, 15 (47%) of 32 risperidone patients but only seven (21%) of 34 haloperidol-treated subjects were so classified (χ
2=5.13, df=1, p=0.02). At the end of the flexible-dose phase, the comparable figures were 61% (N=17) among risperidone patients and 37% (N=11) among haloperidol-treated patients (χ
2=3.35, df=1, p=0.07).
Clinical improvement was analyzed with the same criteria used by Kane et al.
(18). In that study, improvement was defined as improvement in BPRS scores of 20% or more and a CGI severity score of, at most, less than 3 (mild) or a total score of 35 or less on the 18-item BPRS. At the end of the fixed-dose phase, 19% (N=6) of risperidone-treated patients and 3% (N=1) of haloperidol-treated patients met these criteria (χ
2=3.67, df=1, p=0.06). At the end of the flexible-treatment phase, 32% (N=9) of risperidone-treated patients and 14% (N=4) of haloperidol-treated patients met the Kane et al. criteria (χ
2=2.5, df=1, p=0.11).
Risperidone-treated individuals received less concomitant anticholinergic medication than haloperidol-treated patients during both the fixed phases (χ2=11.49, df=1, p=0.001) and flexible phases (χ2=8.97, df=1, p=0.003) of treatment. α-Blockers, which were used for the treatment of akathisia, were less often prescribed to risperidone-treated subjects during the flexible phase only (χ2=4.25, df=1, p=0.04). There were no significant differences between groups in the use of benzodiazepines.
There were no statistically significant differences in the clinical assessment of changes from baseline in specific aspects of drug-induced parkinsonism (tremor, rigidity, or bradykinesia). In addition, we did not detect statistically significant differences in the global score on the Simpson-Angus Scale. On the Barnes Akathisia Scale at the end of the flexible-treatment phase (week 8), 24% (N=7) of risperidone-treated patients had observable akathisia, compared to 53% (N=16) of haloperidol-treated patients (χ2=4.12, df=1, p=0.04). Total AIMS scores (sum of choreic scores from the seven body areas) were lower for the risperidone-treated group at the end of the flexible-dose phase (F=7.91, df=1, 55, p=0.0007). The overall severity score from the AIMS was similarly lower in the risperidone-treated subjects at the end of the flexible-dose phase (F=4.85, df=1, 54, p=0.03).
The risperidone group was dichotomized into robust responders (greater than 40% improvement in BPRS score) (N=10) versus all others (less than 40% improvement in BPRS score) (N=23). The clinical predictors of robust response were more severe positive symptoms, greater conceptual disorganization, and less rated depression at baseline. This subgroup also had significantly higher scores than the remainder of the risperidone group at baseline on measures of acute extrapyramidal side effects (Barnes objective akathisia rating; χ2=5.32, df=1, p=0.02) and tardive dyskinesia (AIMS severity; t=2.15, df=30, p=0.04).
DISCUSSION
Our findings suggest that risperidone may be more effective than haloperidol in treatment-refractory schizophrenia. The superior clinical efficacy was present after 4 weeks of fixed-dose treatment. That clinical superiority was not preserved after an additional 4 weeks of blind treatment, during which the medication was flexibly dosed. The lower use of adjunctive anticholinergics and α-blockers and the greater improvement in the ratings of tardive dyskinesia confirm that risperidone has a lower liability for extrapyramidal side effects than does haloperidol.
A limitation of this design is that patients and physicians might be able to guess at drug group assignment because of the need for concomitant extrapyramidal side effect medication. However, we chose a dose of haloperidol that we thought would minimize this risk. In addition, bias is best and most importantly controlled by the random assignment of patients to both groups.
The data harvested from two dissimilar rating instruments (BPRS and CGI) and two different sources (observing physician and self-reporting patient) confirm the superiority of risperidone treatment. The lack of statistical effect in either the positive or negative symptom domain suggests that risperidone’s enhanced symptom reduction power is either inconsistent throughout the group of patients or spread evenly among a variety of symptomatic domains (e.g., psychotic, affective, anxiety, cognitive) for each patient. These two possibilities are not mutually exclusive. Risperidone may, for example, show great positive symptom reduction in one patient but demonstrate only enhanced subjective tolerability (e.g., lower ratings of anxiety, depression, dysphoria) in another. The dichotomization of the risperidone group into null and robust responders suggests that risperidone’s greatest differential clinical impact is in subjects who demonstrate sensitivity to the extrapyramidal liabilities of conventional agents (akathisia and choreiform dyskinesia at baseline), prominent positive symptoms, and very little depression. The predictive validity of such multiple correlational analyses is suspect, but these results do hint that there may be distinct subpopulations of treatment-refractory individuals who derive clinically pertinent (40% improvement) benefit from risperidone. The existence of this subpopulation (approximately 20% of the risperidone group) also supports the hypothesis that risperidone has a somewhat different mechanism of antipsychotic action than haloperidol.
The apparent impersistence of risperidone’s “superiority” during the flexible-dosing phase (weeks 5–8) has a number of possible explanations. The time course of drug response may be slower for haloperidol than it is for risperidone, so that by week 8, haloperidol-treated subjects had caught up with their risperidone-treated counterparts. There is little hypothetical reason or empirical data from previous trials to support this possibility. The pivotal, multicenter trial in historically responsive subjects
(26) indicated that risperidone may, indeed, have a shorter time to clinical effect than haloperidol—a characteristic that if true, could explain our results. It is more likely, though, that the mean improvement of the haloperidol-treated group was “slowed” by the presence of a larger number of nonresponders.
Another potential explanation for our results is that differential dropout in the two groups changed the configuration of the two populations by the study’s conclusion. However, the number of dropouts in this study was small (N=11 of 67); five of five in the haloperidol group and three of six in the risperidone group were dropped because of lack of drug efficacy. It is unlikely that these few subjects could have so substantially shifted the appearance of efficacy.
Alternatively, the dosing that was employed in the flexible phase may have favored haloperidol over risperidone. In general, the clinical behavior of the treating physicians (D.A.W., W.C.W., and B.D.M.) was to increase the dose of each group above that used during the fixed-dosing phase. The average dose of risperidone was 7.5 mg/day (SD=1.9) and of haloperidol was 19.4 mg/day (SD=5.6) during the flexible-dose phase. It is possible that the method employed pushed the risperidone group beyond the most effective dose range. The results of the CGI (but not the BPRS) seem to confirm this possibility; the proportion of improved subjects in the risperidone-treated group fell from 42% to 28% between the end of the fixed- and the flexible-dosing periods. The comparable percentages for the haloperidol group were 17% and 25%. These results could also have been caused by a time-related “dwindling” of efficacy in the risperidone-treated group. This possibility is not supported by the BPRS data, which showed a steady improvement in both groups throughout the 8 weeks of the study. The risperidone group demonstrated more improvement, but it was of statistically insignificant magnitude by week 8. The lone significant result in
Table 2 (p=0.03 for BPRS total scores) at the end of the fixed-dose phase may have been a type I error (Bonferroni adjusted p=0.18). The effects at week 8 were statistically very small (standardized effects generally less than 0.2), and the study was well powered only for large effects (power=80% for d value of 0.74).
The results on the AIMS similarly have several possible explanations. Type I errors again have to be considered, but there is no obvious (at least to us) bias in the design. Most intriguingly, it is possible that risperidone does, indeed, have a greater ability to suppress choreiform movements than haloperidol. If this is true, it is difficult to explain invoking only the hypothesis of D2 blocking potential of the two groups. Because of the doses employed, the haloperidol group was, without doubt, receiving more D2 blockade (and thus, one would expect, more chorea suppression) than the risperidone group. The more tempting and theoretically consistent explanation is that the greater use of anticholinergics in the haloperidol group resulted in a greater chorea-inducing pharmacologic stress in that group. The results in detail, though, do not appear to support this explanation in that the scores in the haloperidol group were unchanged overall from baseline, whereas those in the risperidone group consistently drifted down from the placebo baseline through the end of the flexible-dose phase. Although it is conceptually unsatisfying, an alternative explanation is that it is possible that risperidone has an ability to suppress choreas other than simply through its inherent ability to antagonize D2 receptors.
In summary, this study demonstrated that during the fixed-dose phase, risperidone was more effective than haloperidol for managing patients with treatment-refractory schizophrenia. It also extended and confirmed the prior observation that risperidone is better tolerated than haloperidol in that it has less extrapyramidal toxicity.