Despite the ubiquity and severity of drug-drug interactions, this problem is one of the most poorly recognized and poorly understood issues within clinical medicine (1, 2) . The impact of drug-drug interactions on patient safety is finally being elucidated, and the magnitude of the problem is vast (3, 4) . Detection and anticipation of these interactions is a daunting task, given the breadth of pharmacodynamic and pharmacokinetic variables with which clinicians must grapple. Physicians are finding it increasingly necessary to become acquainted with the workings of the cytochrome P450 system, phase II metabolism, and even mediators of absorption and distribution, such as the P-glycoprotein transporter. It is not surprising that addressing the subtle and complex drug-drug interactions arising from exotic regimens exceeds the capabilities of most clinicians. However, as the following case illustrates, even well-known and commonplace drug-drug interactions may elude detection and produce significant patient morbidity.
Case Description
Chief Complaint
The patient, “Mr. F,” had experienced persistent dizziness, culminating in a recent fall.
Past History
Mr. F was a 76-year-old Caucasian man treated within the Department of Veterans Affairs (VA) hospital system. He had a known long-standing history of hyperlipidemia, reflux esophagitis, and type II diabetes mellitus with neuropathic pain in both feet. He had smoked one pack of cigarettes per day for the past 55 years. He was in his usual state of health until he was referred to the VA dementia clinic because of declining self-care and noted difficulties in following medical instructions. His prescribed medications included gabapentin, 800 mg t.i.d.; nortriptyline, 25 mg h.s.; enalapril; omeprazole; gemfibrozil; and rosiglitazone. He was also taking docusate sodium, pyridoxine, and multivitamins. He was only intermittently compliant with this regimen, even though he claimed that the gabapentin provided significant relief of his neuropathic pain.
History of Present Illness
During the dementia clinic evaluation, Mr. F was diagnosed with dementia, not otherwise specified; his score on the Mini-Mental State Examination was 23 out of 30. He had specific mild to moderate impairments in memory, visual-spatial functions, and executive functions. During the mental status examination, he was also noted to display rapid and pressured speech, flight of ideas, grandiosity, paranoid ideation, and hypersexuality. Specifically, he stated that he had invented the name for a major food company but that his wife and sister had stolen his ideas and sold them to a rival company, enabling them to make “millions” that were rightfully his. He claimed that this was the main issue that had led to his divorce approximately 20 years ago.
Over the ensuing weeks, family members provided collateral history. Mr. F’s parents actually were quite wealthy and had given him a great deal of money over the years, but he had consistently gambled it away. He had eight children, although he remained in contact with only two of them. His family reported that Mr. F had been persistently emotionally and physically abusive to his wife, which had led to both his divorce and his estrangement from his older children. His family specifically denied his accusations that his wife and sister had tried to steal his business ideas. He did apparently work with various food companies but usually as a blue-collar worker, not a businessman. His family also reported that he had had one prior admission to one of the local state mental health inpatient facilities in the remote past, although details of his diagnosis and treatment were not known. They additionally reported that over the years Mr. F had experienced several discrete episodes characterized by elation, agitation, “hyperactivity,” and decreased need for sleep.
This history supported the diagnosis of bipolar I disorder. The treatment team discontinued his nortriptyline and decided to start treatment with divalproex sodium, 250 mg/day. This was titrated to a total dose of 750 mg/day. A total valproate blood level obtained at that dose was 13.5 ng/ml (therapeutic level, 50–100 ng/ml). Soon thereafter, 325 mg/day of aspirin was added to his regimen as a cardioprotective agent. Since his grandiose and paranoid thought content was directed only toward past circumstances and he displayed no other active psychotic symptoms, the decision was made to not initiate treatment with an antipsychotic agent.
Several months later, Mr. F severely injured his right foot after stepping on a piece of glass; this accident ultimately resulted in rehospitalization. Another psychiatric consultation was obtained for follow-up recommendations. At that time, he was still receiving divalproex sodium, 750 mg/day, and aspirin, 325 mg/day, and his total valproate blood level was 19.3 ng/ml. The consulting psychiatrist suggested further titration of the divalproex dose to produce a total serum trough level of 50–70 ng/ml.
Several weeks later, the hospital pharmacist was formally consulted on the case because of the extensive medication list. Mr. F was now requiring narcotic analgesia for relief of his peripheral neuropathic pain. His medication list included aspirin, 325 mg/day; divalproex sodium, 750 mg/day; oxycodone, 5 mg b.i.d.; fosinopril; gemfibrozil; omeprazole; glyburide; insulin (regular human on a sliding scale); and a multivitamin. The results of pertinent laboratory studies to date included an estimated creatinine clearance of 53 ml/min, a thyroid-stimulating hormone measurement of 2.31 μU/ml, a vitamin B 12 level of 464 pg/ml, a folate level of 16.1 ng/ml, and a nonreactive rapid plasma reagin test. His weight was 89 kg (196 lb). A computed tomography scan of the brain, without contrast, showed only atrophy consistent with the diagnosis of dementia. The pharmacist repeated the psychiatric consultant’s recommendation to increase the divalproex dose. Two weeks later, a new consulting psychiatrist met with Mr. F. This consultant also recommended increasing the valproate dose so as to produce a total valproate blood level of 50–70 ng/ml. Over the next 2 months, the valproate dose was gradually titrated from 750 mg/day to 2500 mg/day. Total valproate blood levels during this period ranged from 16 to 65 ng/ml, and plasma albumin levels during this interval ranged from 1.9 to 2.9 g/dl.
Two weeks after the valproate dose was titrated to 2500 mg/day, Mr. F was noted to be having increasing difficulty with transfers from the bed to a wheelchair, even though he was healing well. Three weeks later, he attempted to transfer to a standing position, became dizzy, was unable to accurately grab for a support, and experienced a painful fall. His difficulties with transfers steadily increased. Another pharmacy review 2 days later suggested only additional oxycodone as needed for breakthrough pain. Soon thereafter, Mr. F was briefly transferred to the acute care VA facility for a preoperative evaluation before excision of a basal cell carcinoma from the left temporal area. At this time, we became aware of the patient’s history, and on the basis of a suspicion of an interaction between aspirin and valproate, we arranged for total and free valproate levels to be measured.
Subsequent Hospital Course and Treatment Recommendations
Mr. F was still receiving divalproex sodium, 2500 mg/day, and aspirin, 325 mg/day. His trough total valproate level was 64.0 ng/ml, and a trough free valproate level was 24.7 ng/ml (therapeutic range, 4.8–17.3 ng/ml), with an albumin level of 2.8 g/dl. In accordance with standard VA hospital protocol, his aspirin was discontinued 7 days before the scheduled procedure. Even though he continued to receive 2500 mg/day of valproate, total and free trough valproate blood levels measured 5 days after the aspirin was discontinued were 36.0 ng/ml and 3.9 ng/ml, respectively, with an albumin level of 2.5 g/dl. Following acquisition of these results, we communicated with the psychiatric consultant and advised that henceforward the patient’s valproate dose should be titrated by following free valproate concentrations rather than total levels. The treatment team restarted his aspirin regimen after his basal cell excision was completed. Since the free valproate concentration varies linearly with the valproate dose, we specifically recommended tapering the valproate dose to 1250 mg/day in order to yield a free concentration in the range of 10–15 ng/ml and then further adjusting the dose on the basis of free valproate concentrations. Following this decrease in his divalproex dose, Mr. F had no further complaints of dizziness or incoordination. However, he was discharged before follow-up measurements of free and total valproate could be obtained at a dose of 1250 mg/day.
Discussion
This case illustrates the ability of aspirin to increase the free concentration of valproate many-fold, while total valproate levels often do not change appreciably (5 – 7) . The basic significance of this drug-drug interaction is that the free fraction of any drug is the fraction that is available to interact with receptors. It is the pharmacologically active fraction. Commonly obtained valproate blood levels reflect the total concentration, which is the sum of the free, active fraction and the bound, inactive fraction. Thus, elevated free valproate concentrations can often produce clinical valproate toxicity, even in the presence of normal total valproate levels (5 – 7) .
Subtypes of Displacement Drug-Drug Interactions
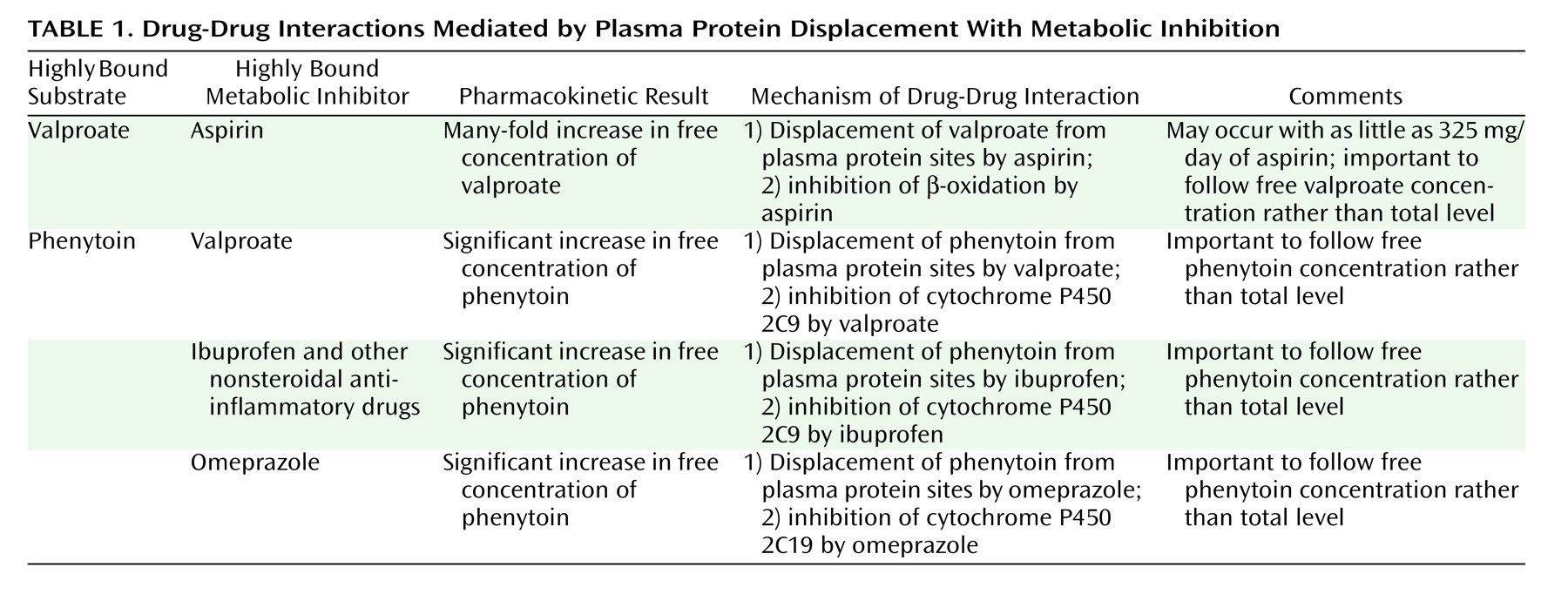
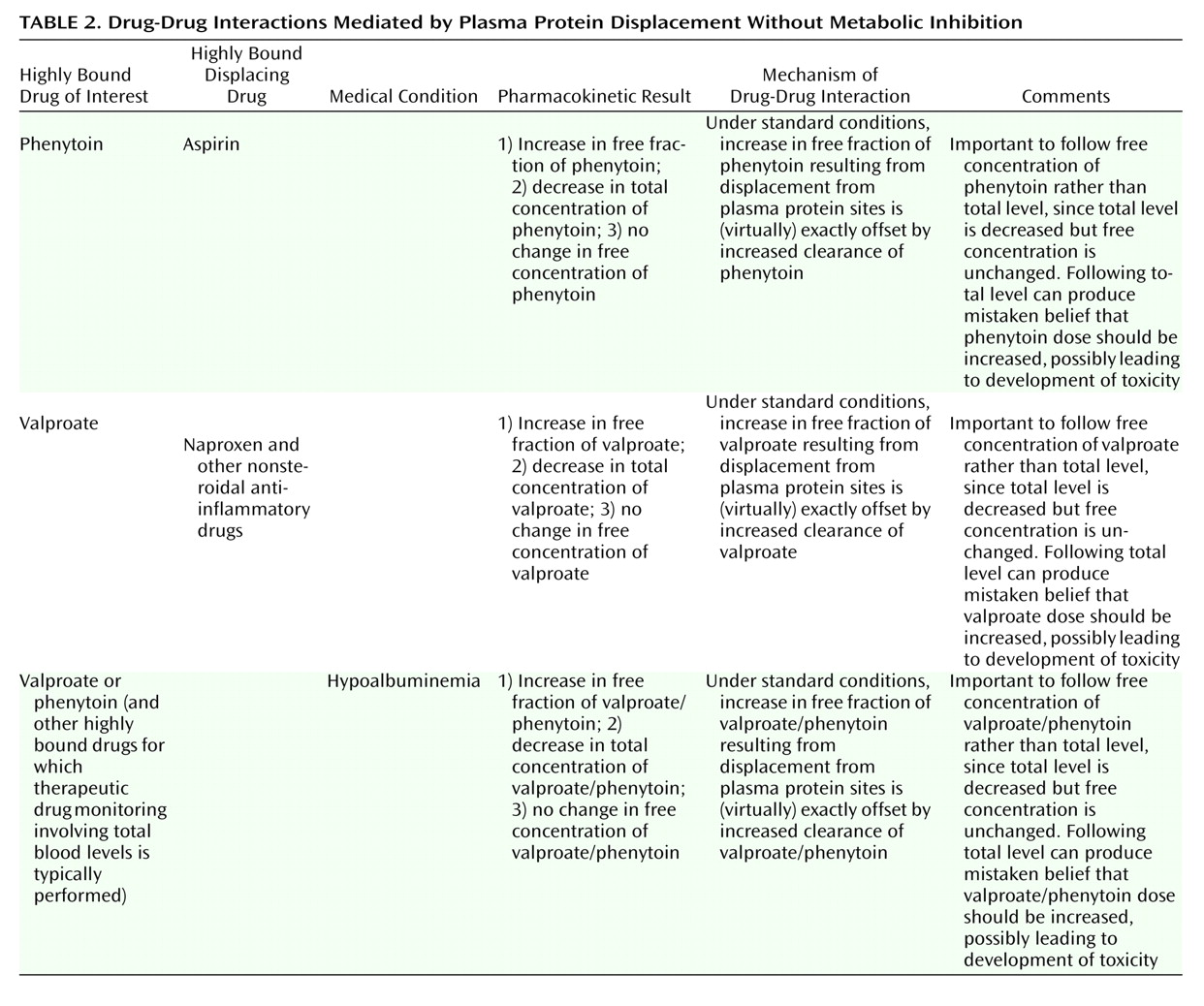
In analyzing this specific interaction, it is helpful to first characterize the various types of clinically meaningful interactions mediated by plasma protein displacement. Broadly, there are two main types of displacement drug-drug interactions: 1) displacement accompanied by metabolic inhibition ( Table 1 ) and 2) pure displacement without accompanying metabolic inhibition ( Table 2 ). Although drug toxicity is the ultimate concern for both types of displacement-mediated drug-drug interactions, the means by which toxicity may arise are quite different.
Displacement Accompanied by Metabolic Inhibition
In the case presented here, aspirin affected valproate levels. The aspirin-valproate interaction is an example of displacement accompanied by metabolic inhibition ( Table 1 ). Both valproate and aspirin are highly bound to plasma proteins, such as albumin (5, 6, 8, 9) . Thus, when they are coadministered at sufficient doses, there is mutual displacement and a rise in the free fraction of each drug. Additionally, aspirin is an inhibitor of beta-oxidation, and this process is responsible for roughly 40% of valproate’s metabolism (5, 6, 8, 9) . These two factors combine to produce modest increases in total valproate levels but disproportionate and often clinically significant increases in free valproate concentrations. The existing literature indicates that combinations of valproate with “antipyretic” doses of aspirin (approximately 3900 mg/day) can produce up to fourfold increases in free valproate concentrations (6) . In the case presented here, the presence of a much smaller dose of aspirin (325 mg/day) produced a more than eightfold greater free valproate level while the total valproate level was only 77% greater with aspirin.
The patient’s low albumin levels may explain part of this apparent disparity between the previous case literature and this case. The lower the albumin level, the more likely it is that a given concentration of aspirin will meaningfully displace valproate from albumin binding sites. Another factor might have been the presence of omeprazole in his regimen. Omeprazole is also a highly bound drug (10), and thus it would have contributed to the displacement of valproate from plasma proteins. These factors would have increased the free fraction of serum valproate. Because this effect is also accompanied by aspirin’s inhibition of beta-oxidation, this increase in the free fraction of valproate will also result in a corresponding increase in the free concentration of valproate.
These factors might account for the magnitude of this disparity. However, another explanation is that aspirin doses significantly lower than “antipyretic” doses of aspirin can produce elevations in free valproate concentrations. To our knowledge, rigorous studies to determine the lowest dose of aspirin that can produce clinically meaningful interactions with valproate have never been performed. Cases such as this suggest that this interaction may well be more prevalent and serious than has been supposed to date.
An oddity of this case was the fact that the patient’s free valproate concentration was only 3.9 ng/ml while he was taking 2500 mg/day of valproate and no aspirin. The patient received all of his scheduled doses of the medication, so noncompliance was not a factor. While this is a much lower value than would generally be expected, the fact that the discontinuation of aspirin produced a disproportionately greater decline in his free valproate concentration than in his total concentration nonetheless implicates the presence of the aspirin-valproate interaction as a significant contributor to his state of valproate toxicity.
The most clinically relevant issues raised by this case are the great frequency and generally unrecognized severity of this drug-drug interaction. Valproate is often used in the treatment of seizure disorders, mood and psychotic disorders, and an array of impulse control disorders and related conditions. Aspirin is widely used in patients with coronary artery disease, to prevent cerebrovascular accidents, as prophylaxis for carcinoma of the bowel, and for an array of other uses. These various conditions are frequently comorbid, resulting in frequent coadministration of aspirin and valproate. Standard practice in maintenance treatment of a patient taking valproate is to follow total valproate concentrations. However, the therapeutic range for total valproate concentrations is predicated on a predictable numerical relationship between total and free valproate concentrations during standard conditions, which include normal albumin levels and an absence of displacement of valproate from plasma protein binding sites by other drugs. When aspirin and valproate are coadministered, aspirin increases the ratio of free valproate concentration to total valproate concentration at a given dose, through the mechanisms we have just listed. Thus, in the presence of aspirin, the titration of valproate dosing based on total concentration, rather than free concentration, runs the risk of producing clinical valproate toxicity. We hope that this case will raise awareness of this drug-drug interaction and the advisability of following free valproate levels with this medication combination.
As was previously mentioned, the aspirin-valproate interaction illustrates the situation when a combination of plasma binding displacement and metabolic inhibition produces increases in the free concentration of a drug (valproate). Another example of this kind of drug-drug interaction would be the impact of valproate on the free phenytoin concentration ( Table 1 ). As with the valproate-aspirin combination, there is mutual plasma protein displacement. Additionally, valproate is an inhibitor of cytochrome P450 2C9 (11), which is the major enzyme responsible for the metabolism of phenytoin (12, 13) . Thus, this combination produces elevations in the free concentration of phenytoin (14, 15) .
In a related vein, with this combination of phenytoin and valproate, phenytoin will actually act to decrease valproate concentrations (8) . This occurs through phenytoin’s induction of cytochrome P450 2C9 and several phase II enzymes that are responsible for much of valproate’s metabolism (16 – 19) . Since the free fraction of valproate is the portion of the total concentration that is available for clearance, then it should be theoretically true that the free concentration of valproate should be disproportionately decreased relative to the decrease in total valproate concentration. However, this dimension of interactions involving plasma protein displacement has not been well characterized or quantified to date.
Displacement Without Metabolic Inhibition
In the other basic type of plasma protein displacement interaction, displacement is not accompanied by metabolic inhibition. An example of this would be the effect of aspirin on phenytoin levels ( Table 2 ). Since the displacement from plasma protein binding sites between aspirin and phenytoin is mutual, there are elevations in the free fraction of both drugs. However, this free fraction, in addition to being the pharmacologically active fraction of the drug, is also the fraction that is available for clearance. Thus, since aspirin does not in any way inhibit the metabolism of phenytoin, there is increased clearance of the free fraction, which compensates for the increased ratio of free to bound drug that is produced by the reciprocal displacement. The net result of these processes is that once equilibrium is achieved, the free fraction of phenytoin remains elevated, the free concentration is unchanged, and the total concentration actually decreases (20, 21) . Another example of displacement without accompanying metabolic inhibition is the effect of other nonsteroidal anti-inflammatory drugs (NSAIDs), such as ibuprofen and naproxen, on valproate ( Table 2 ). NSAIDs naturally displace valproate from binding sites, but unlike aspirin, these other NSAIDS do not meaningfully inhibit the metabolism of valproate. Thus, the free fraction of valproate increases and the total concentration of valproate decreases, but the free concentration of valproate does not change.
Although not really a drug-drug interaction, the situation produced by the presence or development of hypoalbuminemia is exactly analogous to a pure displacement drug-drug interaction ( Table 2 ). Hypoalbuminemia can result from malnutrition, diabetes mellitus, hepatic and renal disease, burns, and pregnancy (22) . If a patient is already stabilized with an appropriate dose of phenytoin or valproate and then develops significant hypoalbuminemia, ensuing displacement of these drugs from plasma binding sites will lead to increases in the free fractions and decreases in the total concentrations, but the free concentrations will remain basically unchanged (15, 22) . This profile will be true for any clinical situation that results in increased displacement without any inhibition of drug metabolism.
Clinical Implications
In the case of drug-drug interactions in which displacement is accompanied by metabolic inhibition, the clinical concerns are straightforward. In this case, free concentrations can rise to a far greater extent than total concentrations. Thus, monitoring only total concentrations in this situation runs the risk of failing to detect drug toxicities that often produce adverse clinical outcomes. The benefits of determining and following free concentrations are apparent.
In contrast, it would be tempting to conclude that since pure displacement drug-drug interactions, and analogous situations like hypoalbuminemia, do not alter free concentrations, then there can be no situation in which they become clinically significant and it is therefore completely safe to simply follow total concentrations. However, pure displacement interactions, when they interface with “standard practice,” can pose real clinical problems for patients. To illustrate this concern, let us assume that one starts with a therapeutic dose and blood level (total and/or free) of phenytoin, and then aspirin is added. The addition of aspirin will produce no change in the free phenytoin concentration and thus no phenytoin toxicity (20, 21) . The important point is that the addition of aspirin will lead to a decrease in the total phenytoin level (20, 21), which might tempt the clinician to inappropriately increase the phenytoin dose in order to bring the total level back to its original value. Doing so would produce an apparently therapeutic total phenytoin blood level, but the patient would likely develop clinical toxicity due to a subsequent increase in the free concentration above the therapeutic range. Thus, even in the absence of metabolic inhibition and resulting changes in free concentrations, there is a utility and rationale for following free concentrations when administering two or more drugs that are highly bound to plasma proteins.
In all of these circumstances, following free drug levels provides a means to recognize and possibly avoid adverse clinical sequelae arising from drug-drug interactions mediated by plasma protein displacement. This principle is generalizable to interactions between most drugs highly bound to plasma proteins. Besides aspirin, phenytoin, and valproate, other examples of highly plasma-protein-bound drugs include aripiprazole; buspirone; clozapine; fluoxetine; HMG-CoA reductase inhibitors, or “statins” (except for pravastatin); other NSAIDS, such as ibuprofen and naproxen; omeprazole; paroxetine; propranolol; protease inhibitors; proton pump inhibitors; sertraline; trazodone; tricyclic antidepressants; typical antipsychotics; verapamil; warfarin; and ziprasidone.
In many situations, titrating doses on the basis of total concentrations of even highly bound drugs is safe and reliable. Most young and comparatively healthy patients do not have hypoalbuminemia. Also, many patients do not concurrently take two or more highly plasma-protein-bound drugs. However, in the treatment of medically challenging or malnourished patients with complex medication regimens, dose titration based on free concentrations of these drugs is a prudent measure. Free concentrations are more expensive to measure than total concentrations, but in such situations the benefits of improved patient safety and maximum therapeutic efficacy more than compensate for these costs.
Footnote
Received Jan. 14, 2006; revision received June 21, 2006; accepted July 5, 2006. From the Baltimore Veterans Affairs Medical Center. Address correspondence and reprint requests to Dr. Sandson, Sheppard Pratt Hospital, 6501 North Charles St., Towson, MD 21204; [email protected] (e-mail).Neil B. Sandson, M.D., Catherine Marcucci, M.D., Denis L. Bourke, M.D., and Rosemary Smith-Lamacchia, C.R.N.P., report no competing interests.
References
1.
Grymonpre RE, Mitenko PA, Sitar DS, Aoki FY, Montgomery PR: Drug-associated hospital admissions in older medical patients. J Am Geriatr Soc 1988; 36:1092–1098
Glassman PA, Simon B, Belperio P, Lanto A: Improving recognition of drug interactions: benefits and barriers to using automated drug alerts. Med Care 2002; 40:1161–1171
Einarson TR, Metge CJ, Iskedjian M, Mukherjee J: An examination of the effect of cytochrome P450 drug interactions of hydroxymethylglutaryl-coenzyme A reductase inhibitors on health care utilization: a Canadian population-based study. Clin Ther 2002; 24:2126–2136
Juurlink DN, Mamdani M, Kopp A, Laupacis A, Redelmeier DA: Drug-drug interactions among elderly patients hospitalized for drug toxicity. JAMA 2003; 289:1652–1658
Regardh CG, Gabrielsson M, Hoffman KJ, Lofberg I, Skanberg I: Pharmacokinetics and metabolism of omeprazole in animals and man—an overview. Scand J Gastroenterol Suppl 1985; 108:79–94
Wen X, Wang JS, Kivisto KT, Neuvonen PJ, Backman JT: In vitro evaluation of valproic acid as an inhibitor of human cytochrome P450 isoforms: preferential inhibition of cytochrome P450 2C9 (CYP2C9). Br J Clin Pharmacol 2001; 52:547–553
Mamiya K, Ieiri I, Shimamoto J, Yukawa E, Imai J, Ninomiya H, Yamada H, Otsubo K, Higuchi S, Tashiro N: The effects of genetic polymorphisms of CYP2C9 and CYP2C19 on phenytoin metabolism in Japanese adult patients with epilepsy: studies in stereoselective hydroxylation and population pharmacokinetics. Epilepsia 1998; 39:1317–1323
Ritter JK, Kessler FK, Thompson MT, Grove AD, Auyeung DJ, Fisher RA: Expression and inducibility of the human bilirubin UDP-glucuronosyltransferase UGT1A1 in liver and cultured primary hepatocytes: evidence for both genetic and environmental influences. Hepatology 1999; 30:476–484
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
For more information or tips please see 'Downloading to a citation manager' in the Help menu.
PsychiatryOnline subscription options offer access to the DSM-5-TR® library, books, journals, CME, and patient resources. This all-in-one virtual library provides psychiatrists and mental health professionals with key resources for diagnosis, treatment, research, and professional development.
Need more help? PsychiatryOnline Customer Service may be reached by emailing [email protected] or by calling 800-368-5777 (in the U.S.) or 703-907-7322 (outside the U.S.).
If the address matches an existing account you will receive an email with instructions to retrieve your username
Create a new account
Change Password
Password Changed Successfully
Your password has been changed
Login
Reset password
Can't sign in? Forgot your password?
Enter your email address below and we will send you the reset instructions
If the address matches an existing account you will receive an email with instructions to reset your password.
Change Password
Congrats!
Your Phone has been verified
×
As described within the American Psychiatric Association (APA)'s Privacy Policy and Terms of Use, this website utilizes cookies, including for the purpose of offering an optimal online experience and services tailored to your preferences. Please read the entire Privacy Policy and Terms of Use. By closing this message, browsing this website, continuing the navigation, or otherwise continuing to use the APA's websites, you confirm that you understand and accept the terms of the Privacy Policy and Terms of Use, including the utilization of cookies.