Even a decade ago, we could not have anticipated the transformation in genetics that now shapes our investigations of the etiology of psychiatric disorders. Advances in genomic methods have opened a window of opportunity through which we are continually expanding our understanding of the multiple links between the genetic mechanisms of psychiatric disease and its cognitive and clinical features. Four papers in this issue exemplify the range and potential of approaches to the study of genotype-phenotype associations in psychiatric disorders that originate in childhood. Each paper brings us a little closer to understanding the complex associations between genetic variation, neuroanatomy, and clinical symptomatology in psychiatric disease.
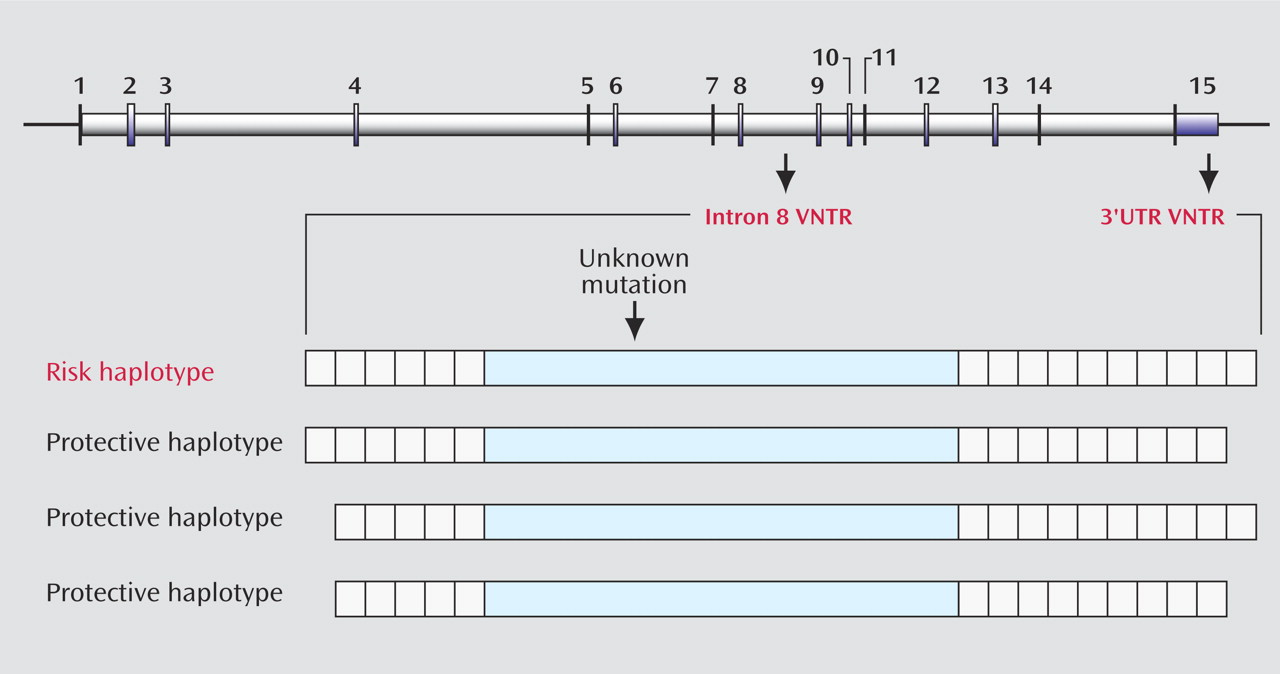
Finding a gene mutation that transmits risk for ADHD is like looking for a needle in a haystack of the 3 billion base pairs of DNA that constitute the human genome. The dopamine transporter gene (DAT1) is one gene that has been strongly implicated in conferring risk for ADHD, but whether it is actually mutated in persons who have ADHD is not certain. An unknown mutation in the gene may have occurred in a single individual many generations ago. In that case, the mutation would be expected to be found in people alive today along with other variations in the gene that were part of the original or founder individual’s genetic makeup. Finding genetic variations that occur more frequently in people who have ADHD than in others could then point to the region of the mutation that actually causes the illness. Asherson and colleagues focus on polymorphisms (i.e., gene variants) located in the untranslated (i.e., non-amino-acid coding) region called 3′UTR and in the intron 8 region of DAT1. Both of these polymorphisms consist of multiple repeating sequences that have no known function but serve to identify the ancestry of a particular individual. In this type of polymorphism, known as a variable-number tandem repeat (VNTR), the number of alleles that make up the polymorphism can vary between individuals. Previous studies have suggested that the 10-repeat allele in the 3′UTR region is associated with risk for ADHD, but these studies have not been consistently replicated. Asherson et al. seek to resolve these discrepancies by showing that the presence of the 10-repeat allele in the 3′UTR region of DAT1 is robustly associated with ADHD only when this allele is found in combination with the six-repeat allele of the neighboring polymorphism in intron 8.
Figure 1 illustrates the different chromosomal patterns that increase or decrease the risk of ADHD. Thus, the paper from Asherson and colleagues further defines the ancestral chromosome that may have contained the genetic mutation in DAT1 and, by replicating previous findings, strengthens the evidence that a genetic change in the synthesis of the dopamine transporter may be part of the mechanism of ADHD.
Identifying the genes that increase the risk for autism is as daunting as the search for ADHD-related genes. Classic autism is part of a spectrum of disorders that overlap in symptomatology and level of impairment. Efforts have been hampered by the complexity of the disorder: autism is defined by a confluence of social impairments, language deficits, and repetitive behaviors, each of which may involve different (but potentially interacting) disease genes. Duvall and colleagues address this issue by using two separate approaches to identify susceptibility genes for autism. In one approach, the authors used a traditional genetic linkage design to examine the transmission of genetic markers from parents to fully affected sibling pairs. In an innovative second approach, they investigated whether genetic markers were associated with variation along a specific endophenotype for autism. An endophenotype is a measurable quantitative trait that is closely linked to the etiology of the illness and is influenced by the genetic risk factors that influence the disease itself. Duvall et al. sought to determine whether genetic markers were associated with varying degrees of impairment in social relatedness, an endophenotype they measured with the Social Responsiveness Scale (SRS), which they administered to all members of each family. The authors found that the use of quantitative data on social relatedness from all members of a family (regardless of the presence of a categorical diagnosis of autism) provided greater statistical power than the traditional linkage analysis to identify associations between genes and behavior.
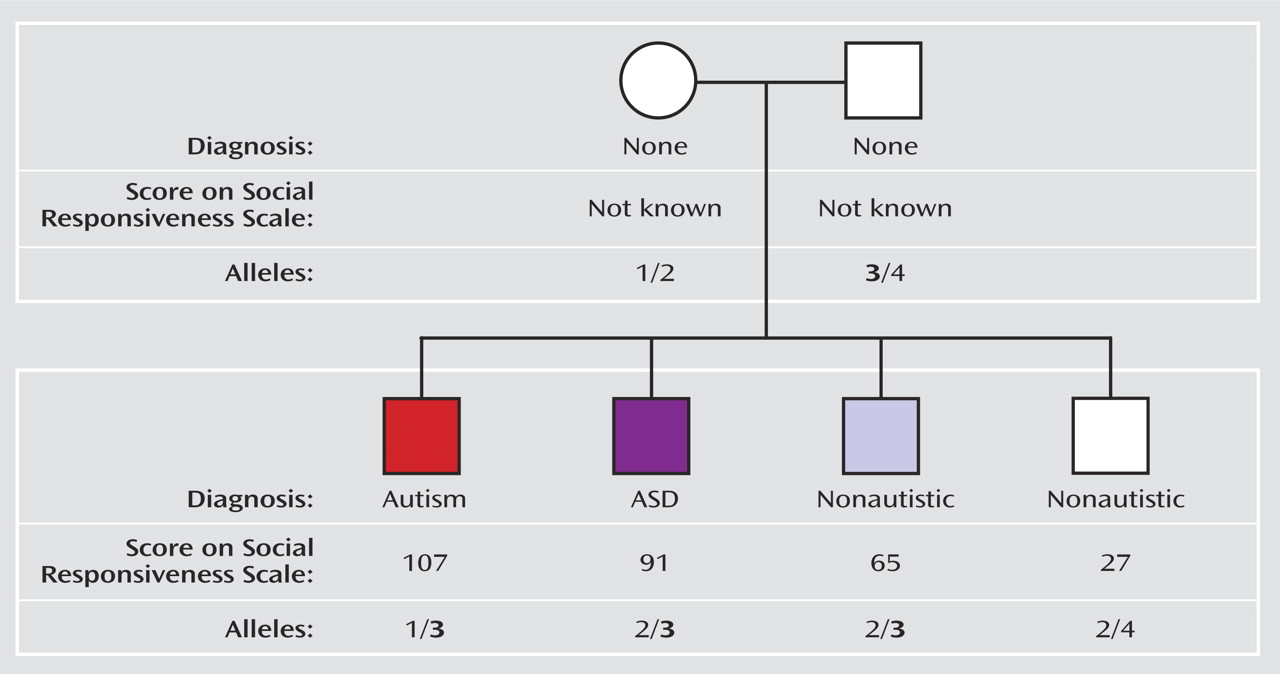
Figure 2, for example, depicts the pedigree of a representative family in the SRS quantitative genome scan study. Categorical diagnoses (autism, autism spectrum disorder, or none), SRS scores, and alleles at the marker showing the strongest linkage signal in the scan are shown for each family member. The nonautistic child with the higher SRS score has an intermediate phenotype that is missed when the categorical diagnoses of autism are used. This child, as well as the child affected with autism or autism spectrum disorder, received allele 3 from the father, but the child with the low, normal-range SRS score did not. Thus, these findings demonstrate the utility and the power of parsing a complex, heterogeneous disorder such as autism into its component endophenotypes and of using a specific quantitative trait to locate susceptibility genes.
Endophenotypes are not limited to behavioral traits. Neuroimaging studies during the past decade have exponentially expanded our understanding of the neuroanatomic phenotypes that are associated with several psychiatric disorders. Quantitative studies of neurodevelopment have illuminated the pathophysiology of the neurocognitive deficits and clinical features that are implicated in a variety of psychiatric disorders originating in childhood, including ADHD and autism. In this regard, the cerebellum has been examined extensively, owing to its involvement in motor timing, attention, executive function, and emotion regulation. Mackie and colleagues investigated the longitudinal trajectory of cerebellar volumes in children and adolescents with ADHD and demonstrated an association between clinical outcome and age-related volumetric changes in the cerebellum. They used scores on the Children’s Global Assessment Scale to divide subjects with ADHD, each of whom had three neuroanatomic magnetic resonance scans acquired about 2.5 years apart, into those with better and worse clinical outcome. As
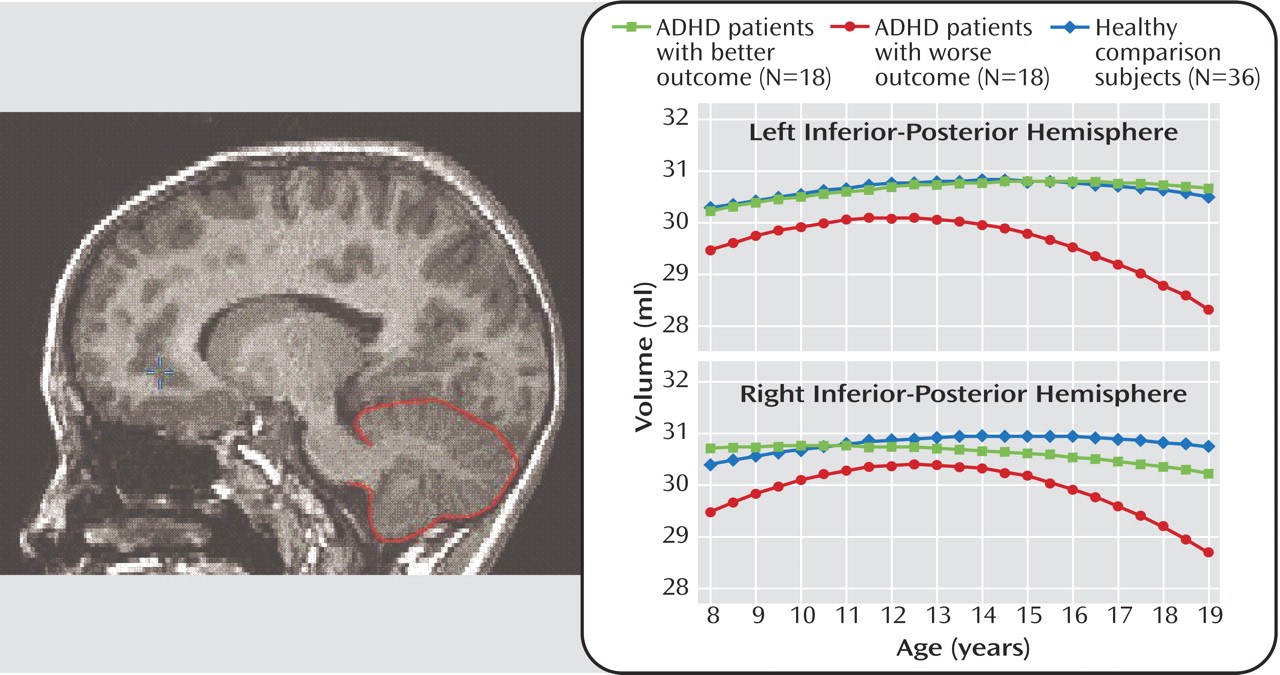
Figure 3 shows, in subjects with ADHD who had a worse clinical outcome, the volumes of the right and left inferior-posterior lobes of the cerebellar hemispheres became progressively smaller during adolescence relative to both healthy comparison subjects and subjects with ADHD who had a better outcome. The volumes of the cerebellar vermis, by contrast, did not show a differential trajectory; they were smaller in both ADHD groups relative to healthy comparison subjects at all time points. Mackie et al. hypothesize that fixed deficits in the cerebellar vermis may constitute an enduring phenotype of ADHD, whereas progressive changes in the cerebellar hemispheres may reflect neuroanatomic plasticity associated with changes in symptoms.
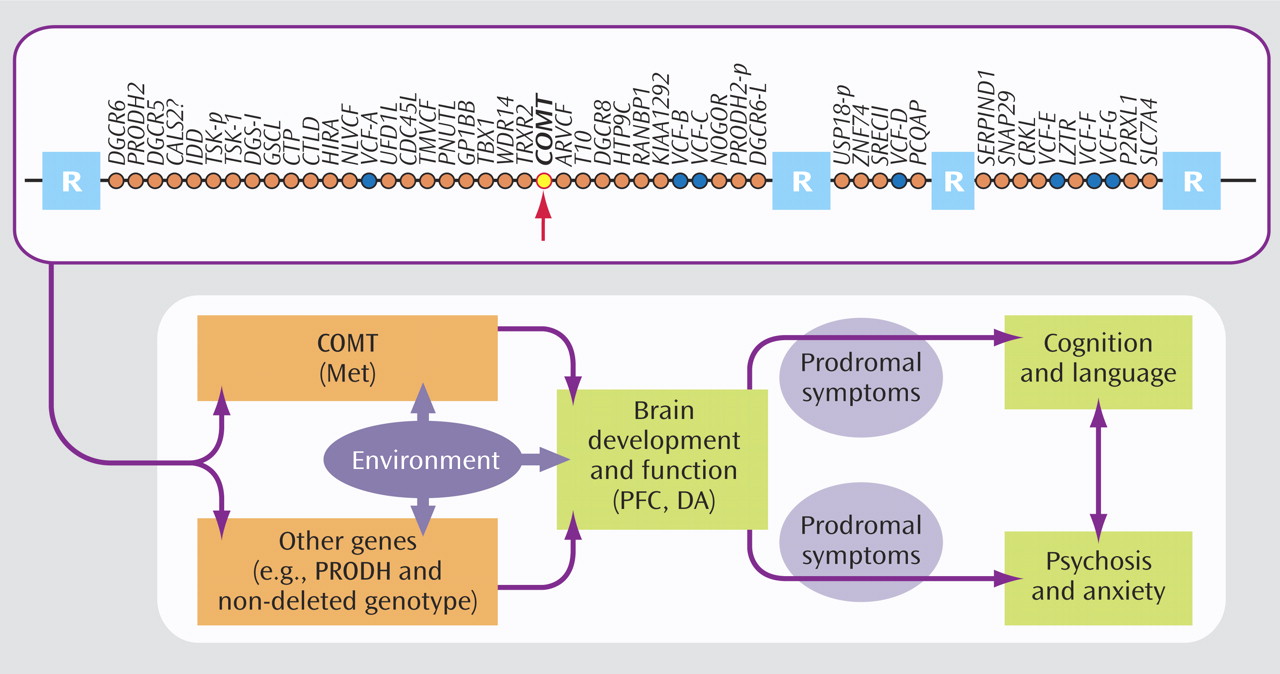
As in ADHD and autism, phenotypic heterogeneity has also complicated the search for candidate genes in psychotic disorders. The 22q11.2 deletion syndrome, caused by a microdeletion of approximately 40 genes (
Figure 4 ) on one copy of chromosome 22, represents an intriguing, etiologically homogeneous model for understanding the mechanisms of psychosis. Up to 30% of individuals with 22q11.2 deletion syndrome (also known as velocardiofacial syndrome) develop schizophrenia-like psychosis in young adulthood. Gothelf and colleagues investigated the genetic, neuroanatomic, and psychiatric risk factors for psychosis in a longitudinal study of adolescents with 22q11.2 deletion syndrome. They focused on the catechol
O -methyltransferase (COMT) gene, which has been implicated in schizophrenia by virtue of its role in the degradation of dopamine in the prefrontal cortex. A well-studied variant of the COMT gene is the polymorphism resulting from a substitution of the amino acid methionine (Met) for that of valine (Val) at a specific location (codon 158) of the gene. The Met allele results in slower breakdown and greater availability of dopamine in the prefrontal cortex. A gene-dosage effect of this polymorphism has been reported in typical individuals: those with two Met alleles perform better on prefrontal cognitive tasks than those with a Met allele and a Val allele, who in turn perform better than those with two Val alleles. The presence of two Val alleles also increases the risk for schizophrenia in some typical individuals. Individuals with 22q11.2 deletion syndrome have either a single Met allele or a single Val allele, since the COMT gene resides in the region that is deleted on one copy of chromosome 22. Gothelf et al. found that the presence of the Met allele, coupled with baseline symptoms of psychosis and anxiety or depression, was significantly associated with the presence of psychosis at follow-up. As
Figure 4 suggests, additional genes may be interacting with COMT to confer risk for psychosis. Nonetheless, these findings are consistent with the notion (1, 2) that relative to individuals with 22q11.2 deletion syndrome who harbor a single Val allele, those with a single Met allele have higher, less optimal levels of prefrontal dopamine, which has a negative effect on their cognitive function and potentially increases their risk for psychosis. Gothelf et al. conclude that the presence of internalizing symptoms or subthreshold symptoms of psychosis in young adolescents with 22q11.2 deletion syndrome may warrant early psychopharmacological intervention to reduce the risk of onset of psychotic illness.
These four papers exemplify divergent but equally valid approaches to the challenges inherent in identifying the genetic mechanisms and pathophysiology of several child psychiatric disorders. Such investigations are often hampered by several factors, including 1) the multifactorial nature of psychiatric disorders, leading to very small effects of several genes across the genome; 2) the complexity and heterogeneity of the clinical phenotypes in a given disorder across individual development and populations of patients; and 3) the evidence that disease genes encode traits associated with the disease rather than the disease itself. Asherson et al. approached this challenge by investigating interactions between genetic variants at different locations of the dopamine transporter gene, thereby strengthening the association between variation of the DAT1 gene and ADHD. Duvall et al. and Mackie et al. demonstrated the power of examining intermediate, quantifiable behavioral and neuroanatomic phenotypes in the search for genotype-phenotype associations in autism and ADHD, respectively. Gothelf et al. illustrated the utility of representing a disorder of known genetic etiology—in this case, 22q11.2 deletion syndrome—as a model for a complex, multifactorial disease such as schizophrenia. Moreover, the papers by Mackie et al. and Gothelf et al. underscore the benefits of using a longitudinal design to model developmental change in clinical and neuroanatomic phenotypes. All of these papers provide new insights into the confluence of genetic and environmental contributions to the etiology and pathophysiology of child psychiatric disorders. Ultimately, these insights should lead to the earlier identification of psychiatric disease and to the implementation of increasingly individualized treatments, reducing the toll these diseases take on families and society.