Antidepressant use by children and adolescents dramatically increased in recent decades
(1,
2), with up to 8 million prescriptions written annually in the United States
(3) . However, the use of antidepressant drug treatment has been fraught with controversy because of questions regarding both efficacy and safety. Efficacy results from pediatric trials are mixed and difficult to interpret, largely because of methodological limitations and regulatory idiosyncrasies in determining what is an “effective” study
(4 –
6) . Furthermore, regulatory agencies in the United States and the United Kingdom raised concerns in 2003 about the emergence of suicidal thoughts or behaviors during antidepressant treatment in pediatric populations, which may have led to a recent decline in prescription rates
(7,
8), rendering risk-benefit analyses even more challenging.
To evaluate the potential association between suicidality and antidepressants, the Food and Drug Administration (FDA) decided to undertake a meta-analysis to examine suicidal events from 24 randomized placebo-controlled pediatric trials of selective serotonin reuptake inhibitors (SSRIs) and other newer generation antidepressants. However, inconsistent labeling of potentially suicidal events was identified as a significant threat to accurate risk-assessment analyses. This concern first arose during an FDA review of one pediatric SSRI study, in which events suggestive of suicidality were labeled “emotional lability.” Subsequent examination of suicidality data from the other eight pediatric antidepressant studies underscored the problem, with a notable example being a subject who slapped herself in the face and was deemed as having made a suicide attempt (
Table 1 ). The FDA determined that conclusions based on these data would be unreliable and might produce either a false signal that would result in unwarranted restriction of useful medications or an underestimation of risk and subsequent danger to the general public.
The problem of inconsistent nomenclature of suicidal ideation and behavior (suicidality) encountered in this data set is not unique. Indeed, the ongoing debate concerning nomenclature has perpetuated the use of multiple terms to refer to the same behavior, frequently with pejorative connotations (e.g., threat, gesture) and descriptors (e.g., “manipulative,” “hostile,” “nonserious”)
(9 –
12) . Such variability in terminology has consequences that extend beyond imprecise communication, limiting comparison of epidemiological prevalence rates and hampering prevention efforts
(13) . Additionally, it undermines the validity of risk-benefit analyses.
To enhance interpretability of pediatric antidepressant trial data to be used in their risk analysis, the FDA commissioned a study by Columbia University/New York State Psychiatric Institute investigators to classify all events that could represent suicidality. The investigators developed a systematic approach to the categorization of potential suicidal adverse events covering the full spectrum of suicidality, rooted in consensus recommendations and empirical findings regarding suicide-related definitions
(10,
12,
14 –
16) .
The whole continuum of suicidality was included in the system, given evidence that manifestations along the spectrum are linked
(17,
18) . For example, evidence suggests that suicide attempts with intent to die are predictive of completed suicide
(16,
18,
19), and individuals who engage in preparatory suicidal behaviors with intent to die are also at risk for future suicide attempts
(20) and completion
(21) . Epidemiological and clinical studies of adolescents and adults have established that severe or pervasive suicidal ideation is a predictor of both future attempts
(17,
22 –
25) and completed suicide
(26) . Moreover, Brown et al. identified passive thoughts about wanting to be dead as a risk factor for completed suicide
(27) . These studies provide the links between manifestations of suicidal process despite well-documented differences between them
(28) .
In the present article, we describe the structure and reliability of the Columbia Classification Algorithm of Suicide Assessment (C-CASA), the classification system of suicidal adverse events that produced the data used by the FDA in their critical assessment of pharmacologic risk.
Method
C-CASA
The C-CASA is a classification system that utilizes definitions of suicidality derived from empirical findings on the phenomenology of suicidality and identified predictive and risk factors. The criteria for a suicide attempt include both self-injurious behavior and suicidal intent (at least some intention to commit suicide). Intent to die portends a risk for future suicide and repeated attempts
(15,
18,
29,
30) and can be reliably obtained
(27) . Inclusion of intent in the definition of suicide allows a distinction between those who self-injure in an attempt to die and those who self-injure for purely other nonsuicidal reasons (e.g., to manage affect)
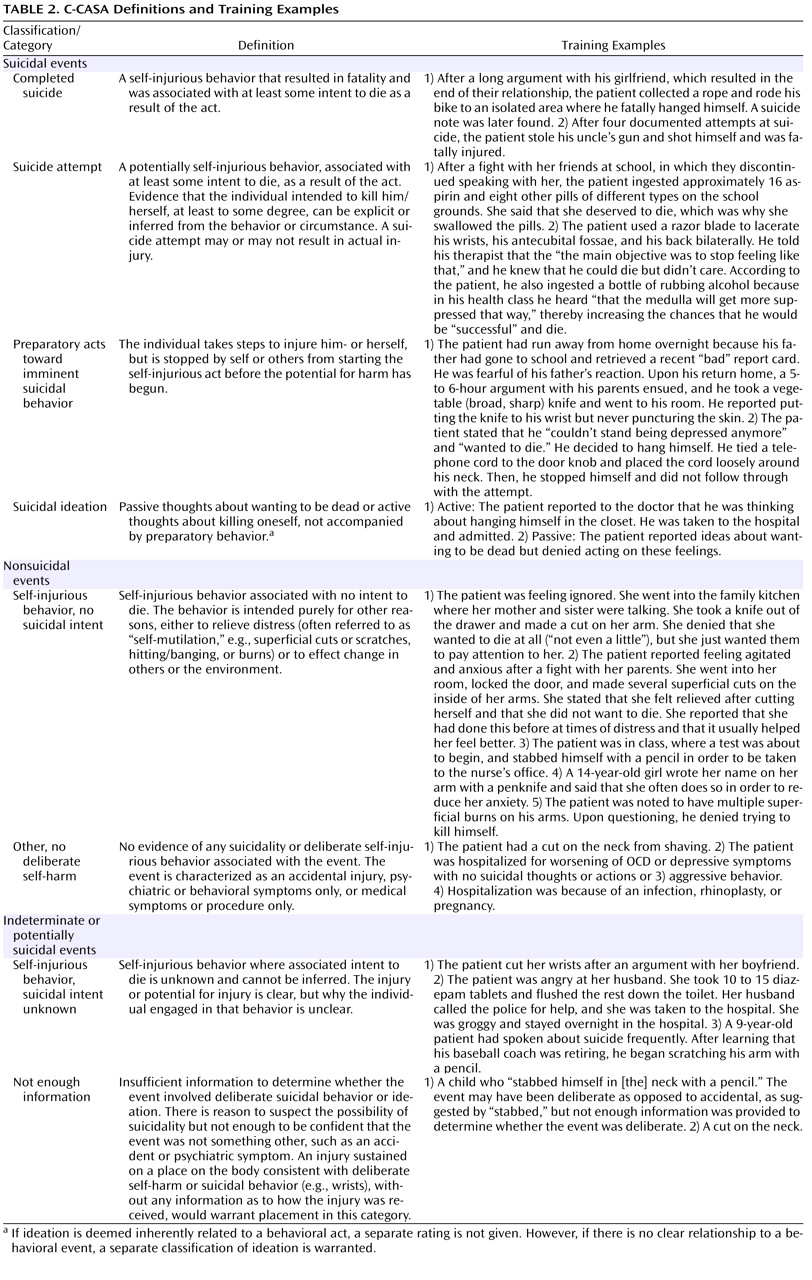
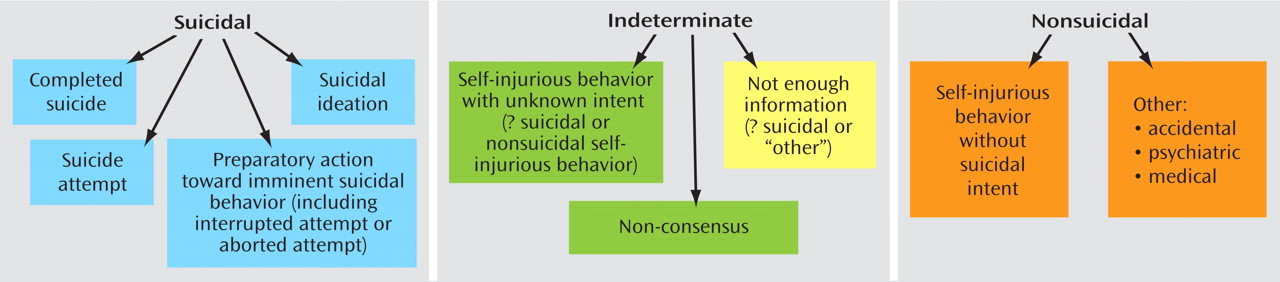
(31) . The C-CASA has eight categories that distinguish suicidal events from nonsuicidal events and indeterminate or potentially suicidal events (
Table 2 ). C-CASA definitions and training examples are presented in
Table 2 .
Figure 1 illustrates the boundaries between categories.
C-CASA Rating Guidelines
The C-CASA includes operationalized guidelines for inference of suicidal intent. “Clinically impressive” behavior or circumstances are used to infer suicidal intent when the stated intent is missing, unclear, or denied. For example, a highly lethal act that is clearly not an accident might mean that no other intent except suicide can be inferred (e.g., a gunshot to the head, jumping from a high-story building). An illustrative example was a case of self-immolation, which was a circumstance allowing inference of intent to classify the event a suicide attempt. Alternatively, inference of suicidal intent could also be based on two other pieces of data, including clinical circumstances such as the method used, number of pills ingested, and location of injury on the body. For example, cuts on the legs typically represent nonsuicidal self-injurious behavior. According to C-CASA guidelines, other relevant data that could be used included past history of suicide attempt, past history of self-injurious behavior/self-mutilation, and family history of suicide/suicide attempts.
Data
Adverse event reports from 25 trials of antidepressant medications with a combined sample of 4,562 pediatric patients were included. Reports were provided by the FDA. Twenty-four trials were sponsored by pharmaceutical companies, and one was funded by the National Institute of Mental Health (NIMH)
(32) ; however, data from that particular trial was subsequently utilized for a pediatric indication by a pharmaceutical company. Twenty-three trials were randomized controlled trials, and two were nonrandomized controlled trials. Participants were pediatric patients, ages 6 to 17 years, and clinical trials were conducted between 1983 and 2004. The treatment duration, across nine medications, ranged between 4 and 16 weeks. Among SSRI-medication trials, two were on citalopram, three on fluoxetine, one on fluvoxamine, six on paroxetine, and three on sertraline. Other newer generation antidepressants studies were three bupropion trials, one mirtazapine study, two nefazodone trials, and four venlafaxine trials. Psychiatric diagnoses treated were major depressive disorder (15 trials), obsessive-compulsive disorder ([OCD] five trials), generalized anxiety disorder (two trials), social phobia (one trial), and attention deficit hyperactivity disorder ([ADHD] two trials). Fifteen of the trials were conducted exclusively in the United States. The two nonrandomized controlled trials were 1) an open-label trial of bupropion for ADHD (N=17) and 2) a randomized withdrawal study of paroxetine for OCD (N=194). The FDA analysis
(33) used a subset of events, classified by the C-CASA, from the 23 randomized controlled trials described previously and events from an additional federally funded trial (Treatment for Adolescent Depression Study). Events from the Treatment for Adolescent Depression Study were classified using the C-CASA but were not included in the present reliability study, since a different pool of raters was used and it was sponsored by NIMH.
Adverse Events
Pharmaceutical company identification of “possibly suicidal” events
The FDA requested that manufacturers of all nine antidepressants identify adverse events that could represent “possibly suicidal” events. Events were identified using an electronic text-string search of trial databases of patient data recorded by local study clinicians. Pharmaceutical companies were asked to search for any adverse events report that included the terms “suic overdos attempt,” “cut,” “gas,” “hang,” “hung,” “jump,” “mutilate,” “overdos,” “self-damage,” “self-harm,” “self-inflict,” “self-injur,” “shoot,” “slash” in the labeling of an event. The FDA permitted exclusion of obvious false positives (e.g., “gas” in “gastrointestinal”). The pharmaceutical companies were also asked to select a subset of events that were considered suicide attempts. No definitional criteria were given to categorize possibly suicidal events and suicide attempts. The string search identified 114 possibly suicidal events; of these, 87 (76.3%) were considered suicide attempts by pharmaceutical companies.
Broadening of event search
To insure that all potentially suicidal events were identified, the scope of the search was broadened beyond those events originally identified by pharmaceutical companies to include all accidental injuries, overdoses, and serious adverse events, such as life-threatening events and hospitalizations. Inclusion of these additional events enabled a blinded review, since both suicidal and other adverse events were included. For classification, 427 potentially suicidal adverse events were included. Among these events, 114 were originally rated by pharmaceutical companies as possibly suicidal.
Adverse event narrative construction
Once adverse events were flagged by the string search, pharmaceutical companies composed narratives for each adverse event using data from case report forms, recorded by local study investigators during the course of the trials, and other sources, such as hospital records. When available, narratives included age, sex, history of suicidality, hospitalization status, current psychosocial stressors, and family history of suicide.
Blinding
Columbia University investigators developed comprehensive blinding procedures that removed information from all narratives that might have biased a classification decision. The FDA then implemented these procedures, removing all potential drug-identifying information, including the drug name, company/sponsor name, patient identification numbers, primary diagnosis, active or placebo arm, and all medication names and types, since treatment with other medications may be associated with a particular antidepressant side-effect profile. Case numbers that had no link to patient identifying information were randomly assigned to narratives by the FDA. Columbia University investigators further removed all original labels given by the pharmaceutical companies to categorize events (“preferred terms”) as well as adverse event labels given by participating investigators, including “serious” and “nonserious” determinations.
Expert Raters
Nine internationally recognized experts in suicide and suicide assessment were recruited as “raters.” Expert review of cases was needed for inference of suicidal intent based on the details of behaviors and related clinical data, since many narratives lacked stated suicidal intent. Expertise in suicidality was determined by relevant experience and publications. Panel members neither were involved in these industry trials nor were employed by Columbia University.
Randomization and Expert Review Procedures
Event narratives were randomly distributed among raters using a balanced incomplete block design. Each event was classified by three raters; each triad of raters shared five cases. This randomization approach reduces rater burden without sacrificing precision in variance estimates
(34) .
Raters participated in a training teleconference to review classification parameters (categories, associated definitions, and case examples), followed by training reliability exercises prior to receiving narratives. Training exercises of each rater were reviewed for agreement with C-CASA definitions, and disagreements were discussed with the individual rater.
Each rater classified approximately 125 events. Raters could consult with a Columbia University trainer regarding the application of classification processes but were restricted from discussing specific events. Cases with discordant ratings were identified, and corresponding narratives were resent to raters. If ratings did not result in a unanimous agreement, a consensus discussion including the three raters assigned to assess the event was held and was led by another rater. The goal was to reach 100% agreement; otherwise, the event was classified as “indeterminate.” Final consensus classification determinations were provided to the FDA.
FDA Independent Audit of the C-CASA
To assess the reproducibility and reliability of the C-CASA methodology, four independent, nonsuicidologist FDA clinical reviewers were selected, including two pediatricians, one pharmacist, and one psychiatrist. Fifteen percent of the 427 event narratives were selected for review, with oversampling of “difficult-to-classify” cases. Raters received the same training and procedures as the expert panel. Audit results showed 89% agreement (kappa=0.84) between audit ratings and expert ratings
(35) .
Statistical Analysis
Reliability coefficients were estimated with a random-effects linear model using the restricted maximum likelihood algorithm in SPSS 12.0 for Windows. Random effects modeled event-to-event, rater-to-rater, and error variation. Intraclass correlation coefficients (ICC) were estimated by the ratio of the variance because of the event divided by the total variance (sum of event-to-event, rater-to-rater, and error variation)
(34) . ICCs were estimated for each category.
Cohen’s kappa was used to evaluate the agreement between pharmaceutical companies and C-CASA classifications. These analyses were conducted with only one event per subject. For subjects with multiple events, statistical calculations used the most severe event, which was chosen according to the severity hierarchy employed by the FDA for their unblinded analyses. This severity hierarchy was as follows: suicide attempt>preparatory behavior>suicidal ideation>self-injurious behavior intent unknown>not enough information>self-injurious behavior, no suicidal intent. This approach identified 377 individual subjects, all of whom experienced one or more relevant adverse event. Only 50 individuals had more than one event, and most of those were accidental injuries.
Blinded examination of de-identified case records was considered exempt from review by the institutional review board of the New York State Psychiatric Institute and the Columbia University Department of Psychiatry.
Results
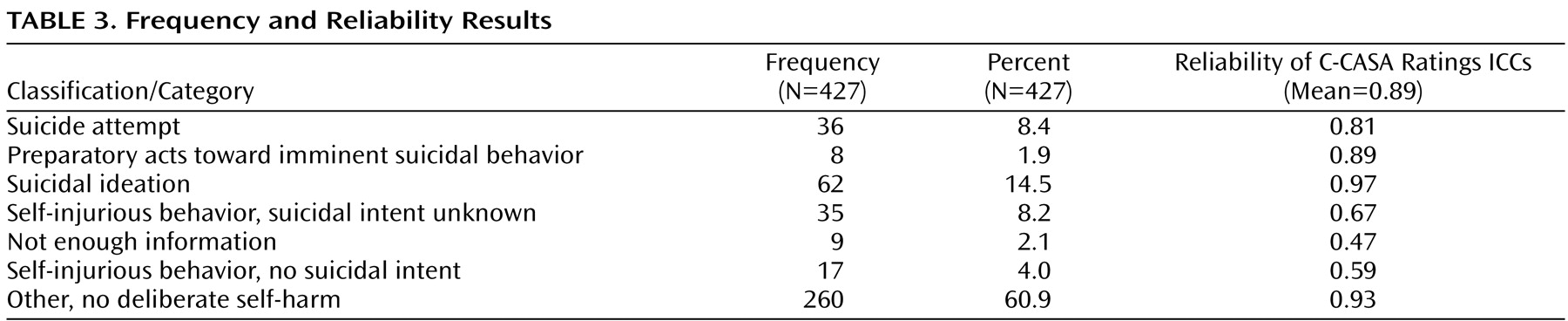
Frequencies of the 427 events according to C-CASA classifications are presented in
Table 3 . Completed suicides are not included, since none occurred in the pediatric trials.
Reliability of C-CASA
Excellent overall reliability (median ICC=0.89) was demonstrated among independent ratings of nine experts using the C-CASA. ICCs for the seven categories are presented in
Table 3 .
Of the 427 events, 366 (85.7%) had unanimous agreement among the three raters. Fifty-nine events (13.8%) had agreement between two of three raters, while two (0.47%) events had no agreement. Consensus discussions were held via teleconference whereby agreement was reached for all cases that were not unanimous.
Comparison With Pharmaceutical Companies
Discrepant cases
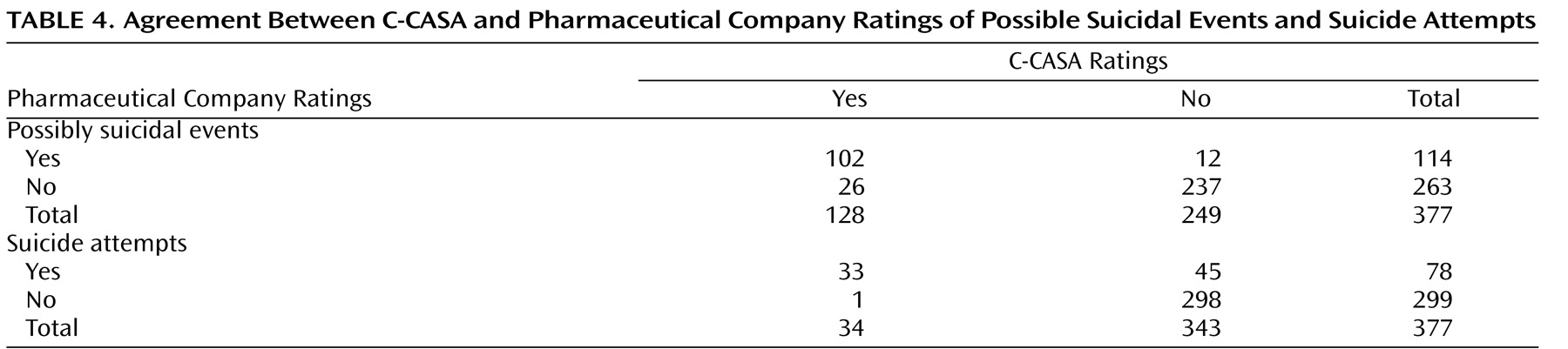
Thirty-eight discrepant cases were identified when comparing C-CASA with pharmaceutical company ratings (
Table 4 ). Of these, 26 were new, possibly suicidal cases that were originally labeled by pharmaceutical companies as something other than suicidal (e.g., accidental injury). These cases were as follows: one suicide attempt, one suicidal preparatory act, 13 suicidal ideation events, four self-injurious behaviors with unknown intent, and seven cases without enough information but reason to suspect suicidality. The following is an example of a newly identified suicidal event: “The patient, age 11, held a knife to his wrist and threatened to harm himself. The patient was hospitalized with an acute exacerbation of major depressive disorder.” The original adverse event label was “exacerbation of major depressive disorder,” without an indication of suicidality from either the site investigator or pharmaceutical company. The new label was preparatory suicidal behavior. This event was discovered only because it was within a serious adverse event report of a hospitalization.
Twelve cases that were originally identified as potentially suicidal by pharmaceutical companies were classified as not potentially suicidal by C-CASA raters. These events were reclassified as psychiatric, involving no suicidality (N=2), accidental injury (N=1), and self-injurious behavior without suicidal intent (N=9).
Agreement on suicide attempts
Modest agreement was found between pharmaceutical company and C-CASA raters’ classification of suicide attempts (kappa=0.53 [SE=0.06]) (
Table 4 ). Of their 114 possibly suicide-related events, pharmaceutical companies rated 78 (68.4%) as attempts, versus the C-CASA raters identifying 34 out of 128 (26.6%) as attempts. Forty-five of the 78 (57.7%) events classified as suicide attempts by the pharmaceutical company raters were not classified by C-CASA raters as suicide attempts. One suicide attempt was identified by C-CASA raters that had not been identified by pharmaceutical companies. Although the C-CASA identified more potentially suicidal cases overall, the rate of specific suicide attempts was lower.
Agreement on definitely suicidal cases
Agreement between C-CASA and pharmaceutical company ratings increased when comparing the broader C-CASA categorization of definitely suicidal events (attempts, preparatory acts, and suicidal ideation) with the pharmaceutical company rating of possibly suicidal cases (kappa=0.69 [SE=0.04]). Thirty-two events identified as possibly suicidal by pharmaceutical companies were not classified as definitely suicidal by the C-CASA. Conversely, 15 newly identified definitely suicidal cases were identified by the C-CASA. This C-CASA grouping was used by the FDA in their primary analysis
(33) .
Agreement on possibly suicidal cases
When comparing the broad nonspecific pooling of all categories that could possibly represent suicidality, there was good agreement between C-CASA (suicide attempts, preparatory behaviors, suicidal ideation, self-injurious behavior with unknown intent, and not enough information) and pharmaceutical company identification of possibly suicidal events (kappa=0.77 [SE=0.04]) (
Table 4 ). This C-CASA grouping was used in the FDA’s “sensitivity analysis” to conservatively examine results that included anything that could have possibly represented suicidality (i.e., “worst case”)
(33) . Thus, the C-CASA identified an increased number of possibly suicidal events in the data set overall.
Discussion
Classification of suicidal adverse events in 25 pediatric antidepressant trials with the C-CASA resulted in reliable classification of suicidal events. The C-CASA classification identified 38 discrepant cases, including events not previously deemed potentially suicidal (N=26) and those changed from suicidal to nonsuicidal (N=12). Furthermore, while C-CASA classification found more suicidal events, estimates of suicide attempts were significantly reduced. The new potentially suicidal events identified involved both suicidal ideation and behavior, across a range of classifications. Thus, when we expanded the search, many new suicidal events were found that had been missed by the pharmaceutical companies. However, of the suicidal events that the pharmaceutical companies identified, C-CASA classification resulted in a 50% reduction in the rate of suicide attempts. This reflects a tendency of the pharmaceutical companies to label any potentially suicidal event or self-injurious behavior as a suicide attempt (e.g., suicidal ideation or a “slap in the face” labeled suicide attempt). These findings underscore the need for a standardized assessment of suicidality. Additionally, the need to expand the search for suicidal events as evidenced by the 26 newly found cases suggests that approaches currently employed in clinical trials lack sensitivity.
When comparing the C-CASA ratings with pharmaceutical company ratings, a relatively low level of agreement was found with more specific identification of suicidal occurrences, namely suicide attempts. Only when identifying a “suicidal range” or a broad nonspecific category of “possibly suicidal” was there better agreement. Pharmaceutical companies rated 45 events as suicide attempts that C-CASA raters did not. Thus, with respect to suicide attempts, reclassification with C-CASA would yield less of a hazard from the medication than if the original pharmaceutical ratings were used. Indeed, the FDA safety analysis that used these C-CASA ratings
(33) found reduced risk estimates of suicidality in a depressed pediatric sample when compared with earlier FDA estimates that relied on the pharmaceutical labels
(36) . Additionally, a more precise risk estimate resulted (i.e., tighter confidence interval) using the C-CASA. These findings support the notion that misclassification may lead to overestimation of true risk
(37) . Such a change in risk estimation has clinical implications and likely affects risk-benefit analyses. Furthermore, the final FDA data set with the C-CASA ratings
(33) included one-third (38/114) of cases that were different compared with the original data set
(36), a substantially different sample. The use of data sets with imprecisely classified suicidal events can result in misleading findings, such as inaccurate risk and protective factors for suicidality.
The reliability of this classification approach was confirmed by the FDA’s independent audit, which concluded that the C-CASA was “robust and reproducible”
(35) . The reliable use of this classification schema by nonsuicidologists reflects the transportability of this methodology. Notably, the FDA has mandated application of C-CASA to classify suicidal adverse events in adult antidepressant trials, as well as nonpsychotropic drug classes, and other centrally acting agents, including all anticonvulsants, cannabinoid 1 receptor (CB1R) inverse agonists for the treatment of obesity and metabolic disease. C-CASA classified data were used in the recent FDA investigation of an association between antidepressants and suicidality in adults
(38) .
Limitations and Future Directions
The study findings are limited by the quality of the available data describing adverse events. Descriptions of suicidal occurrences were variable and limited, particularly regarding intent. Furthermore, the expanded search for unidentified occurrences elucidated the inadequate quality of the elicitation and description of suicidal adverse events.
Although neither the C-CASA raters nor Columbia University investigators were responsible for subsequent analysis using C-CASA ratings—by Hammad et al.
(33) in the FDA’s safety analysis, for example—some discussion of the limitations of these subsequent analyses is warranted. Suicidal adverse events were not systematically elicited but were revealed spontaneously, allowing the possibility of ascertainment bias. Subjects receiving active medication may be more likely to report suicidal occurrences than those on placebo because of increased contact with providers, consequent to other side effects. Such ascertainment bias is an alternate explanation for differential rates among subjects receiving drug treatment versus those receiving placebo found in the FDA safety analysis
(33) . In addition, improvement from active medication may lead subjects to discuss suicidal thoughts with their clinician for the first time, as opposed to such thoughts being caused by the medication.
Future intervention trials that prospectively and systematically monitor occurrence and emergence of suicidality with consistent methods of ascertainment would be informative. Such investigations would more optimally delineate the relationship between suicidal adverse events and antidepressant treatments as well as for any other treatment risk analysis. Improved assessment of suicidal events is necessary both to better inform research-derived risk-benefit analyses and to foster improved clinical management and identification. Accordingly, a prospective counterpart to this system, the Columbia Suicide Severity Rating Scale
(39), is being widely used and frequently recommended by the FDA. The Columbia Suicide Severity Rating Scale is a tool designed to systematically assess and track suicidal adverse events (behavior and ideation) throughout any clinical trial as well as other settings.
The strength of this suicide classification system is, perhaps, in its ability to comprehensively identify suicidal events while limiting the overidentification of suicidal behavior. This classification system is research-based and can be applied in both clinical and research settings. Its use might result in more accurate identification of suicidality and more precise communication among researchers and clinicians, which would ultimately benefit treatment of suicidal individuals. The incorporation of research-supported, standardized suicidality terminology into psychiatric diagnostic manuals could also promote greater accuracy in communication between clinicians, allowing dissemination to a broad audience. Such a common language of suicide classification could be used in the same way that diagnostic criteria are currently used to provide a method for precise, widely understood communication.