Monoamine oxidase inhibitors (MAOIs) were first observed to have mood elevating effects in the early 1950s
(1). Within a few years, several MAOIs were introduced and were soon in wide clinical use
(2). As a result of reports of acute hypertension following the ingestion of dietary tyramine (the “cheese reaction”) and toxic interactions with other medications, coupled with the introduction of tricyclic antidepressants and, more recently, selective serotonin reuptake inhibitors, MAOIs have come to be recommended only for use in major depression with “atypical” features and for treatment-resistant depression
(3).
Because the treatment response to MAOIs is often superior to that obtained with other antidepressants
(4–
7) and these agents may be effective where other treatments have failed
(8,
9), continued efforts have been made to develop MAOIs that do not require restriction of dietary tyramine
(10).
One strategy has been to exploit the existence of multiple isoenzymes of MAO (MAOA and MAOB)
(11). Both isoenzymes oxidize tyramine to inactive metabolites, but it is MAOA, the predominant isoenzyme in the intestinal epithelium and an important component enzyme in the liver, that maintains a barrier to absorption of dietary tyramine
(12). Therefore, selegiline (formerly called
L-deprenyl), which is a selective type B MAOI, has been investigated as a potential antidepressant
(10).
Selegiline ((R)-(-)
N,2-dimethyl-
N-2-propynylphenethylamine HCl) is a highly selective, irreversible MAOB inhibitor at low doses. Several controlled clinical trials have demonstrated its antidepressant activity
(13–
15). However, these trials were conducted at doses sufficient to inhibit both MAOA and MAOB activity, which has been observed to increase tyramine sensitivity
(16,
17). This reflects the important role that MAOA inhibition plays in the antidepressant activity of drugs in this class
(18). It has been a consistent finding that oral selegiline treatment requires dietary tyramine restriction when used in doses sufficient to treat major depression effectively
(10).
The selegiline transdermal system was developed to deliver sustained selegiline blood concentrations without extensive inhibition of intestinal mucosa and liver MAOA
(19). In clinical trials, this novel delivery system has been shown to be devoid of clinically meaningful increases in tyramine sensitivity
(20–
22). Indeed, the transdermal formulation of selegiline induces no greater sensitivity to tyramine than the currently approved oral dose of 10 mg/day used for the treatment of Parkinson’s disease
(23).
We report here the first placebo-controlled trial of a transdermally delivered selective MAOI, the selegiline transdermal system (20 mg applied once daily by means of a 20-cm2 patch), in outpatients with major depression. Four subsequent trials have been conducted; two of these showed the selegiline transdermal system to be more effective than placebo, and two showed no difference between drug and placebo (data on file, Somerset Pharmaceuticals).
Method
A randomized, double-blind, placebo-controlled, fixed-dose, parallel-group trial was conducted at six sites to assess the safety and efficacy of the selegiline transdermal system in adult outpatients with major depression.
Male and female outpatients, 18 to 65 years of age, who met DSM-IV criteria for major depressive disorder, single episode or recurrent, and who had no other primary psychiatric diagnosis were eligible for enrollment. Diagnostic assessment was made using the Structured Clinical Interview for DSM-IV. A score of 20 or higher on the 17-item Hamilton Depression Rating Scale was required at intake; subjects whose Hamilton depression scores decreased by 20% or more or fell below a total score of 20 during the placebo run-in period were dropped from the trial to eliminate early placebo responders.
Exclusion criteria included failure to respond to more than one adequate trial of an approved antidepressant medication for the current episode of depression, any previous manic or hypomanic episode (unless clearly secondary to antidepressant therapy), presence of substance abuse disorder within the previous 6 months, presence of a primary psychiatric illness other than major depression, and exposure to ECT within 90 days of initiation of the trial.
In addition, pregnant and breast-feeding women were excluded, and all women of childbearing age who were included in the study were using an adequate method of birth control. Finally, patients were excluded who displayed any medical illness that could compromise their safety or interfere with implementation of the protocol or interpretation of study results.
All of the patients were free of any psychoactive medications for either five elimination half-lives or 2 weeks before study initiation (whichever was longer) or, in the case of MAOIs, 2 months before study initiation. Sympathomimetic agents were prohibited during the course of the study.
Efficacy Measures
Efficacy was evaluated by using the 17- and 28-item versions of the Hamilton depression scale, the Montgomery-Åsberg Depression Rating Scale, and the Clinical Global Impression (CGI) severity of illness and improvement measures. Additional responder analyses were performed on the intent-to-treat study group with the last observation carried forward. A positive response in Hamilton depression scale scores was defined as either a decrease of 50% or more from baseline or a 17-item Hamilton depression scale endpoint score less than 8. Similarly, a positive response according to CGI improvement score was defined as a change from baseline to much improved or very much improved. The Medex Depression Evaluation Scale, a self-report instrument devised for this study, was used to assess change in sexual function during treatment. The Medex Depression Evaluation Scale measured 1) decreased interest in sex, 2) arousal problems during sex, 3) problems maintaining interest in sex, 4) problems achieving orgasm, and 5) diminished satisfaction from sexual activity. Symptoms were rated from 1 (not at all) to 5 (severe) or 6 (not applicable). Higher scores (exclusive of a score of 6) indicated greater impairment.
Procedures
After a 1-week, single-blind placebo lead-in, subjects were randomly assigned to active treatment or placebo for a 6-week double-blind treatment period. Subjects applied a 20-cm2 patch each morning on the upper body in a relatively hairless area above the elbow; the application site was changed each day. Postbaseline assessments occurred at the end of weeks 1, 2, 3, 4, and 6 of treatment. Medication compliance was documented at each visit by a count of returned patches.
Safety was assessed by physical examination, ECG, and clinical laboratory tests at screening and at endpoint. At each visit, patients’ vital signs were recorded, including three measurements of orthostatic blood pressure and pulse. Subjects were asked about adverse events at each visit and 30 days after the study completion.
Because this was the first large study of the selegiline transdermal system for major depression, subjects followed a tyramine-restricted diet during the trial and for 2 weeks following the end of treatment. Use of medications known to interact adversely with MAOIs was also prohibited. Chloral hydrate, 1000 mg up to four times weekly, was permitted if needed for insomnia but was not allowed the evening before an evaluation.
Written informed consent was obtained from all patients after a complete description of the study was given and before any study procedures were initiated. The study was approved by the institutional review board at each participating site.
Statistical Analysis
A sample size of 176 (88 per group) was calculated as necessary to detect a between-group difference of approximately 3 units in the mean change from baseline in 17-item Hamilton depression scale scores at week 6 with 80% power. We conducted the efficacy analyses on the intent-to-treat patient study group using the last observation carried forward. The intent-to-treat study group included all randomly assigned patients who received selegiline and had an evaluation while they were taking the medication. Continuous data were analyzed by using a two-way analysis of variance (ANOVA); qualitative data were analyzed by using the Cochran-Mantel-Haenszel procedure.
Pretreatment CGI severity of illness ratings were summarized as frequency distributions and analyzed with a center-stratified Cochran-Mantel-Haenszel sum test of mean rank scores. Total scores obtained at baseline from the Hamilton depression scale (1–17 items and 1–28 items) and Montgomery-Åsberg Depression Rating Scale assessments were summarized as averages per treatment group, and changes from baseline were analyzed by using a two-way ANOVA that accounted for study site and treatment group. The Hamilton depression scale item 1 (depressed mood) and item 3 (suicide) ratings were analyzed with a Cochran-Mantel-Haenszel test similar to that used to analyze the CGI severity of illness ratings.
Safety analyses included patients who received at least one dose of selegiline. All adverse events were summarized by using the Coding Symbols for a Thesaurus of Adverse Reaction Terms (COSTART) body system and COSTART preferred term within each treatment group. Fisher’s exact test was used to test for differences between the two groups for the proportion of patients for each reported adverse event and body system. Statistical comparisons between treatment groups for blood pressure and heart rate were made by using a general linear model that considered study site and treatment group. Physical examination and ECG results were compared between treatment groups by using McNemar’s test for categorical data. Clinical laboratory results were summarized as descriptive statistics.
Results in this study were considered statistically significant when the appropriately calculated two-sided p value was ≤0.05. All statistical analyses were performed by using SAS version 6.12 (SAS Institute, Cary, N.C.).
Results
A total of 177 subjects were randomly assigned to the selegiline transdermal system (N=89) or placebo (N=88); 152 (86%) completed the trial. Ten patients (11%) receiving selegiline and 15 patients (17%) receiving placebo dropped out of the study before week 6. Primary reasons for discontinuation were ineffectiveness of the treatment (five selegiline and nine placebo patients), withdrawal of consent (two selegiline and three placebo patients), and adverse events (three selegiline patients, all application site reactions).
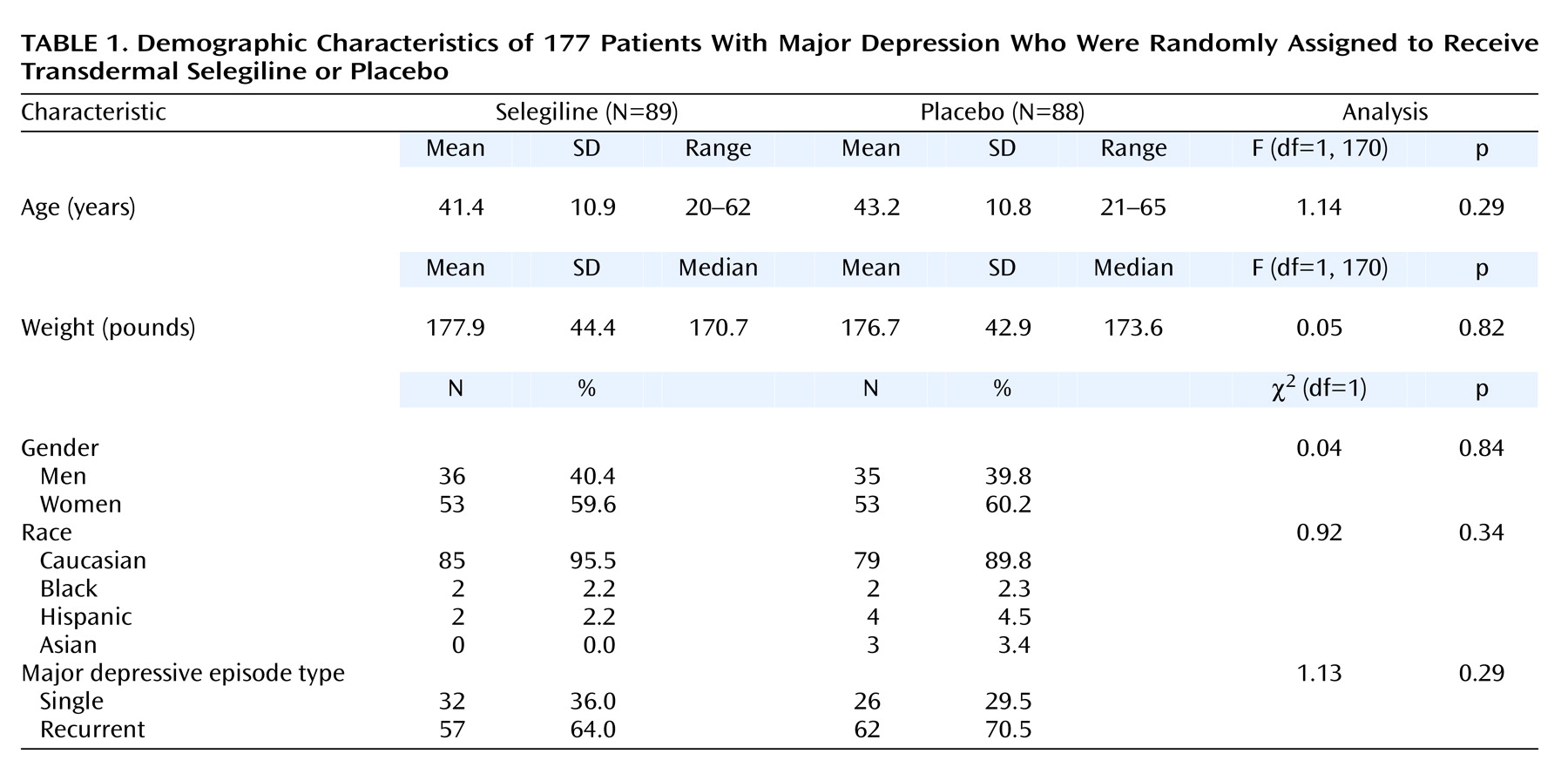
The mean age of the selegiline patients was 41.4 years (SD=10.9); the mean age of the placebo patients was 43.2 (SD=10.8). On hundred six (60%) of the patients were women, and 164 (93%) were white. Most of the patients (N=119 [68%]) had recurrent major depression. No statistically significant differences were noted in the demographics of the two treatment groups (
Table 1). Compliance with study medication averaged 94% (N=166).
Efficacy
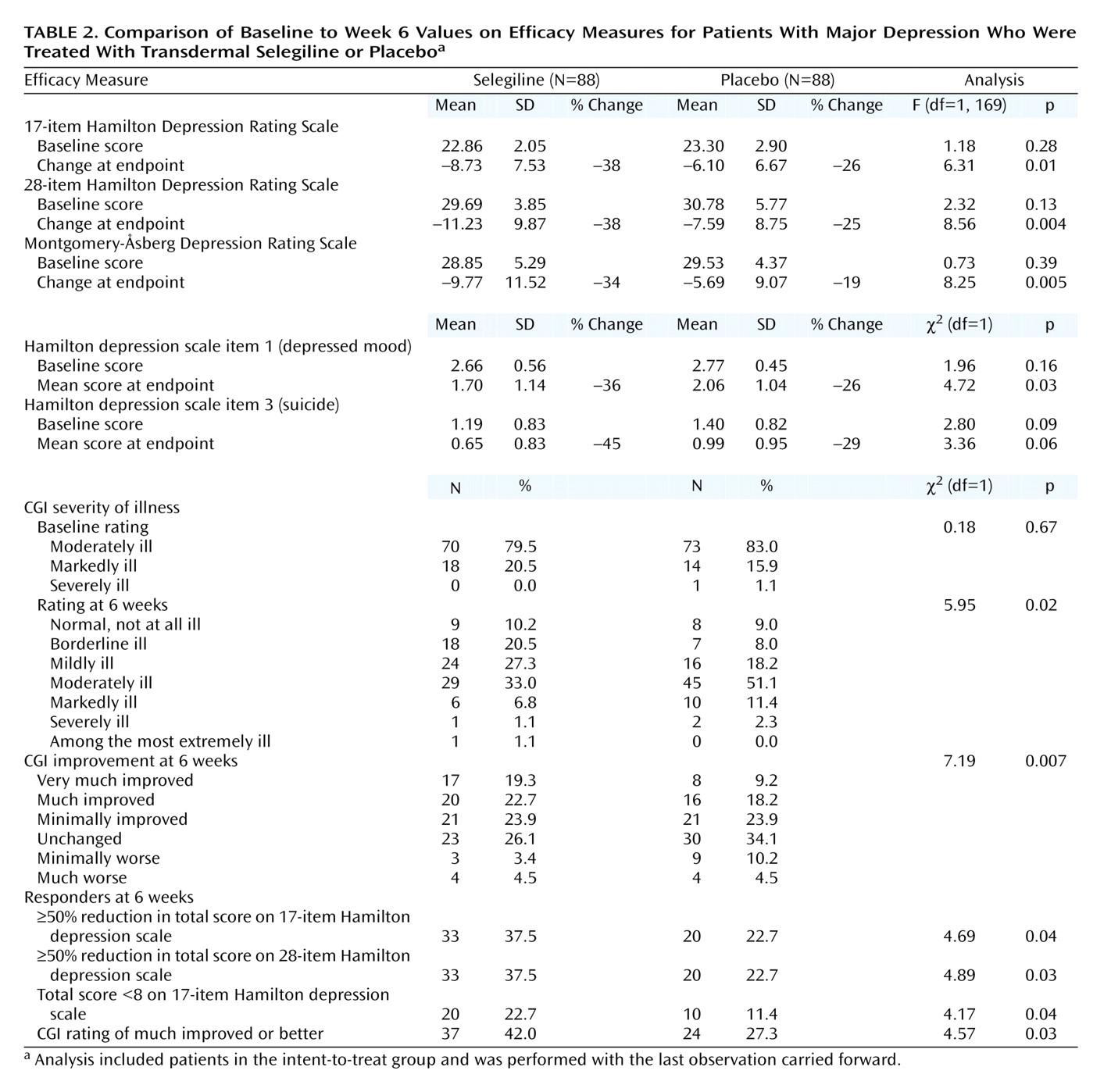
The intent-to-treat efficacy study group consisted of 88 patients in each group (one selegiline-assigned patient did not have an evaluation while receiving the medication). No significant differences between the groups at baseline were noted on any clinical measure (
Table 2). At baseline, the mean 17-item Hamilton depression scale, 28-item Hamilton depression scale, and Montgomery-Åsberg Depression Rating Scale scores were 22.86 (SD=2.05), 29.69 (SD=3.85), and 28.85 (SD=5.29), respectively. Most of the patients were rated as moderately ill on the CGI at baseline.
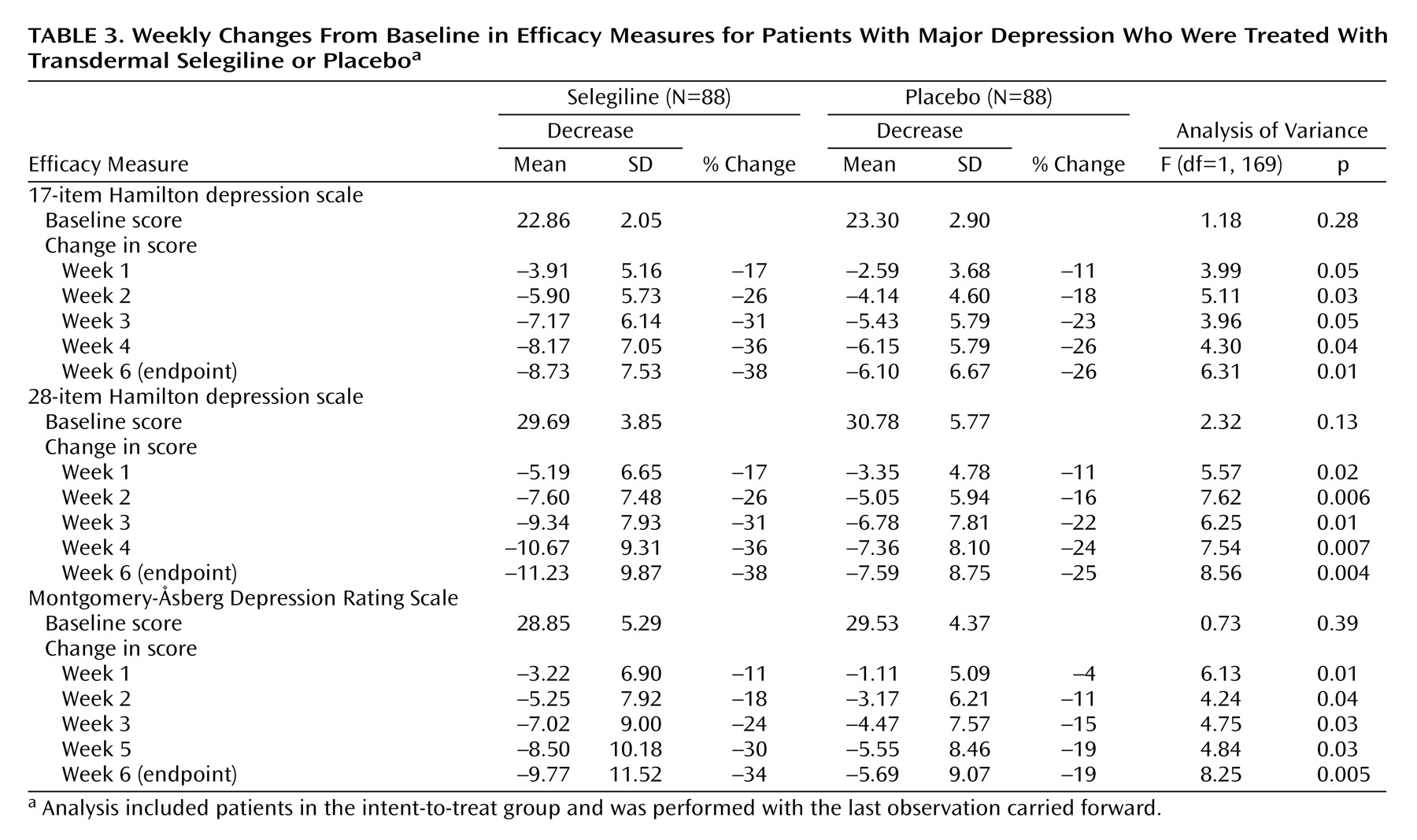
The selegiline transdermal system demonstrated superior efficacy compared with placebo on all clinical measures at endpoint. Forty-six percent greater improvement was seen on the 17-item Hamilton depression scale, 52% on the 28-item Hamilton depression scale, and 79% on the Montgomery-Åsberg Depression Rating Scale. Greater reductions in mean 17-item and 28-item Hamilton depression scale and Montgomery-Åsberg Depression Rating Scale scores were observed with the selegiline transdermal system than placebo as early as week 1 of treatment (
Table 3). The scores of the selegiline patients on Hamilton depression scale item 1 (depressed mood) improved significantly more than those of patients given placebo (
Table 2). The scores of the selegiline patients improved more than those of patients given placebo on Hamilton depression scale item 3 (suicide), but the difference between groups was not significant (
Table 2). CGI ratings showed significantly less severity of illness and greater global improvement in the selegiline group than in the placebo group (
Table 2).
Additional Responder Analyses
Significantly more subjects were considered responders to treatment in the selegiline group than in the placebo group on the basis of changes in Hamilton depression scale scores (
Table 2). A larger percentage of selegiline subjects demonstrated a 50% or greater reduction in 17-item and 28-item Hamilton depression scale total scores (
Table 2). Similarly, a larger percentage of selegiline patients than placebo patients demonstrated remission with a final 17-item Hamilton depression scale total score less than 8 (
Table 2). In addition, robust improvement in the CGI rating (much improved or better) was observed in a greater percentage of selegiline patients than placebo patients (
Table 2).
Safety
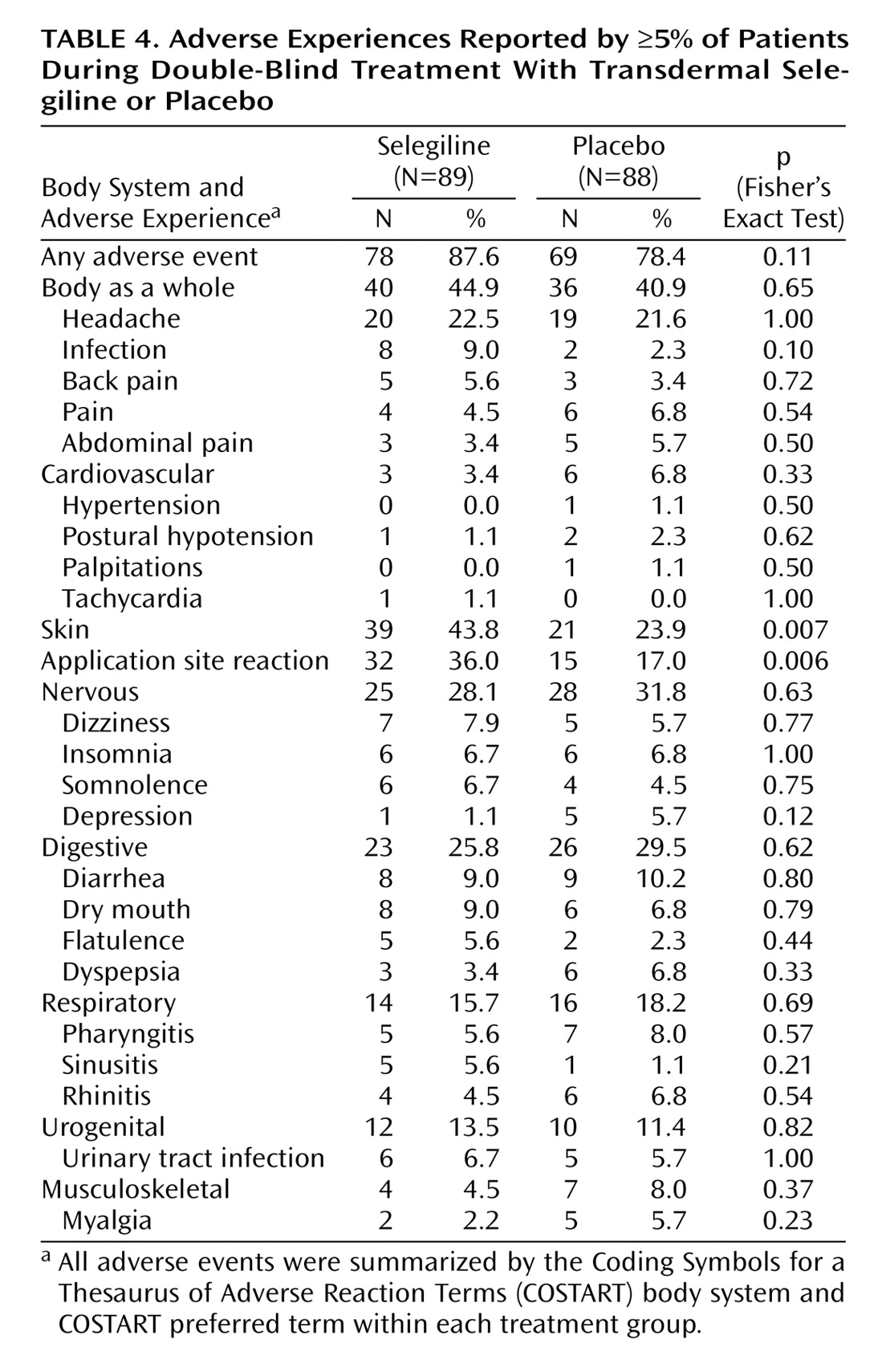
The safety analysis included 89 selegiline and 88 placebo subjects. Overall, treatment was extremely well tolerated: only four patients (4.5%) in the selegiline group and five patients (5.6%) in the placebo group discontinued treatment due to adverse events, including worsening depression. No significant differences between the two groups were noted for adverse events affecting the body as a whole or in cardiovascular, nervous, digestive, respiratory, urogenital, or musculoskeletal systems (
Table 4).
Application site reactions (primarily described as rash, itching, redness, or irritation) were more common in patients treated with the selegiline transdermal system (32 subjects, 36%) than in those given placebo (15 subjects, 17%) (p=0.006, Fisher’s exact test). Five of the 32 selegiline patients required symptomatic treatment for the application-site reactions with topical corticosteroids or oral diphenhydramine, and three subjects in the selegiline group discontinued treatment because of the reaction.
Orthostatic hypotension and other changes in blood pressure following treatment with selegiline were closely monitored. The two treatment groups demonstrated comparable baseline vital signs, although patients in the placebo group had a slightly higher mean standing systolic blood pressure (119 mm Hg) than those given selegiline (114 mm Hg) (F=7.00, df=1, 170, p=0.009). The selegiline group had a slightly greater mean orthostatic change in blood pressure at week 6 (–2.3 mm Hg) than the placebo group (–0.8 mm Hg) (F=15.75, df=1, 170, p=0.0001); however, these orthostatic changes were not considered clinically meaningful. One patient given selegiline (1%) and two patients given placebo (2%) complained of hypotensive symptoms at some time during the study. None of these patients met criteria for orthostatic hypotension on examination.
Ventricular heart rate, sinus rhythm/waveform, PR interval, and QRS and QTc intervals were comparable at baseline in the two treatment groups. The QTc interval at endpoint was slightly decreased in the selegiline group (mean=–0.005 seconds, SD=0.016) but slightly increased in the placebo group (mean=0.001 seconds, SD=0.023) (F=4.27, df=1, 170, p=0.04) at endpoint. No clinically meaningful ECG changes were observed.
Of note, the incidence of cardiovascular adverse events was less than 5% in both treatment groups, and no hypertensive episodes were reported.
Sexual Side Effects
Medex Depression Evaluation Scale scores for both treatment groups were identical at baseline. In the 152 patients completing the trial, the 79 treated with selegiline exhibited significantly improved sexual function (mean=–0.9, SD=6.2) compared with the 73 patients given placebo (mean=1.5, SD=6.8) (F=4.78, df=1, 145, p=0.03).
Discussion
This is the first reported clinical trial of transdermally delivered selegiline for the treatment of depression. Several studies have shown selegiline to be an effective and relatively side-effect-free antidepressant when given in high oral doses requiring restriction of dietary tyramine
(13–
15). We demonstrated that transdermally delivered selegiline, in a dose regimen that appears devoid of clinically significant interaction with dietary tyramine
(20,
23–25), offers significantly better therapeutic benefit than placebo in major depression.
Transdermal delivery of selegiline provides several pharmacological advantages over oral delivery. First, it sufficiently reduces exposure of the gastrointestinal tract to the drug to limit inhibition of intestinal MAOA activity
(21). Thus, adequate gastrointestinal MAOA enzyme is left intact to metabolize dietary tyramine. Second, transdermal administration of selegiline circumvents first-pass hepatic metabolism, which results in sustained high plasma levels of the parent compound with a concomitant decrease in metabolite formation
(26). This provides sufficient brain concentrations of selegiline to produce an antidepressant effect, presumably involving substantial MAOA as well as MAOB inhibition. This also may permit the expression of additional pharmacological properties of selegiline other than MAO inhibition previously observed in vitro
(27). At the same time, there is less exposure to
L-methamphetamine and
L-amphetamine metabolites than observed with oral selegiline
(26).
Because tyramine restrictions were followed in this initial trial, the risk of a “cheese reaction” could not be assessed. However, oral tyramine challenges in normal subjects after fasting
(24) demonstrated that doses of 200 mg or more of oral encapsulated tyramine were required to produce a pressor response. By comparison, when similar tyramine challenges were conducted with tranylcypromine-treated subjects, pressor responses were elicited after 10 mg of oral encapsulated tyramine. The dose of oral tyramine required to produce pressor effects in selegiline patients (200 mg or more) is far in excess of typical dietary intake; a tyramine-rich meal may contain up to 40 mg. Thus, it is unlikely that the selegiline transdermal system will require dietary restrictions.
As in previous studies of selegiline treatment of major depression, the profile of adverse events differed little between the selegiline transdermal system and placebo. This is notable, given that MAOIs, as a class, are liable to have numerous side effects. Of particular clinical importance, no differences from placebo were observed in adverse events related to cardiovascular function, such as flushing, tachycardia, headache, lightheadedness, blood pressure elevation, or orthostatic hypotension. Only skin reactions at the patch site occurred significantly more frequently with the selegiline transdermal system (36% versus 17%) (p=0.006, Fisher’s exact test). This erythematous, occasionally urticarial local reaction generally persisted for several days after each application. Application site reactions occurred in this study at rates comparable to those reported for nicotine patches (34%)
(28). Lower rates have been reported with transdermal estrogen (4%–10%)
(29) and nitroglycerin patches (15%)
(30). It is notable, however, that only three subjects dropped out of the trial because of application site reactions, indicating this was a generally tolerable side effect.
The favorable side effect profile is likely the basis for the unusually high rate of treatment compliance observed in this study. Both treatment groups used virtually all prescribed patches. In addition, 89% of the subjects receiving active drug (79/89) completed the trial. Given the poor rate of treatment adherence generally observed in depressed patients
(31,
32), the high rate seen here may reflect an important therapeutic advantage of the transdermal route of administration.
An intriguing finding was a significant difference between selegiline and placebo at week 1. Although this is not highly unusual in clinical trials of antidepressants, it raises the possibility of an accelerated therapeutic response because of the parenteral drug delivery route, as has recently been demonstrated with intravenous antidepressants
(33). Because of the delayed onset of clinical response to oral antidepressant medications, further investigation of the potential for a more rapid antidepressant response to transdermal treatment is warranted.
Three methodologic limitations must be considered in interpreting these results. First, the 6-week duration of active treatment was relatively brief and may have underestimated the treatment effect of the selegiline transdermal system. Second, the fixed-dose design made it impossible to assess dose-response characteristics of the selegiline transdermal system. Finally, subjects in the present study were prescribed a tyramine-restricted diet. Accordingly, additional studies without dietary restrictions will be necessary.
Conclusions
This is the first reported clinical trial of transdermal selegiline for the treatment of major depression. The selegiline transdermal system was superior to placebo on all measures of efficacy. Transdermal delivery of selegiline has several advantages, including minimal interaction with dietary tyramine, reduced exposure to drug metabolites, sustained exposure to the parent compound, and the possibility of hastened onset of therapeutic effect. In conjunction with its favorable side effect profile, including a paucity of sexual side effects, transdermal selegiline may offer an effective alternative to currently available antidepressants. Additional clinical trials to more fully investigate the characteristics of this new antidepressant treatment are warranted.