Schizophrenia is a complex brain disorder characterized by clinical heterogeneity and neurobiological dysfunction. The etiological importance of genetic transmission in the pathogenesis of schizophrenia has been well established
(1–
4). However, no susceptibility genes have yet been found in investigations using clinical diagnosis as a phenotype, and the lack of consensus in current linkage findings suggests that multiple genes and substantial genetic heterogeneity are likely to be involved in schizophrenia.
One promising strategy that has been advocated for genetic studies of schizophrenia is to broaden the phenotypic characterization of subjects to include discrete neurobiological traits associated with the illness
(5–
9). In addition to their elevated risk for schizophrenia, many first-degree relatives who remain clinically unaffected throughout their lives show impairments in neurobiological functioning similar to those found in schizophrenia patients. These neurobiological disruptions represent an integral part of the pathophysiology of schizophrenia and likely represent more proximal functions of gene action than schizophrenia itself. Therefore, these impairments may be “endophenotypes” for schizophrenia, neurobiological traits that reflect an underlying genetic liability for schizophrenia and can thus identify unaffected relatives who are carriers of a genetic predisposition for the illness (for instance, see references
2 and
10).
In order to qualify as an endophenotype for schizophrenia, a neurobiological abnormality must be heritable and must occur at a significantly higher rate not only in schizophrenia patients but also in their unaffected first-degree relatives than in normal comparison subjects. The higher the relative risk for the abnormality, defined as the ratio of the rate in relatives to the rate in normal subjects, the greater the power to detect genetic linkage
(11–
13). Furthermore, the abnormality should reflect a stable trait that does not fluctuate with clinical state and treatment.
One of the most widely recognized neurobiological abnormalities associated with familial schizophrenia is deficient gating of the auditory event-related potential, a dysfunction in the mechanisms responsible for modulating the brain’s sensitivity to sensory stimuli
(14–
16). The P50 sensory gating paradigm uses EEG techniques to assess an individual’s ability to gate, or inhibit, repetitive auditory stimuli.
The P50 sensory gating deficit has shown strong potential for serving as an endophenotype in genetic studies of schizophrenia
(8,
17). P50 sensory gating deficits have been confirmed repeatedly in schizophrenia patients
(14,
18–29), including patients with illness of recent onset
(30). Furthermore, Siegel et al.
(31) reported that deficits in P50 sensory gating extend to approximately half of the first-degree relatives of schizophrenia patients, indicating a neurobiological trait with a potentially dominant mode of transmission. Impaired P50 sensory gating in relatives was confirmed in other groups of families by the University of Colorado research group
(32,
33) and was independently replicated in a study by Clementz et al.
(29). Heritability of the P50 ratio measure of sensory gating was established by two twin studies
(34,
35).
The presence of the P50 deficit in healthy relatives suggests that the trait is not an artifact of treatment or clinical status, an important characteristic for an endophenotypic trait marker. However, I know of only one study that directly compared P50 sensory gating performance in medicated and unmedicated patients, a more stringent test of medication effects. Freedman et al.
(15) found similar P50 deficits in medicated and unmedicated schizophrenia patients, but the unmedicated group had been free of typical antipsychotics for only a limited time, “at least 10 days.” To my knowledge, no P50 sensory gating studies have been conducted with untreated patients whose illness was in remission. Since the introduction of atypical neuroleptics, research attention has shifted to the effects of these newer medications, and several studies
(30,
36–39) have confirmed that atypical antipsychotics tend to normalize P50 sensory gating in schizophrenia patients. In view of the large proportion of patients now receiving atypical rather than typical antipsychotics, P50 sensory gating studies must now address medication effects, which may complicate the use of the measure as an endophenotypic marker for schizophrenia.
In the Republic of Palau, an isolated island nation in Micronesia, there is a high prevalence of schizophrenia, and cases cluster in large multigenerational families, which are not inbred. The Palau schizophrenia families and the diagnostic criteria used to identify schizophrenia cases have been previously described in detail
(40). Briefly, complete ascertainment of cases and families in Palau revealed 160 narrowly defined cases of schizophrenia in 59 separate families, but 11 of these families had five to 14 cases apiece and represented nearly half of the schizophrenia cases in Palau. The majority of cases could be linked together into extended pedigrees with complex multilineal patterns of disease inheritance.
Palau offers unique advantages for studying P50 sensory gating as a potential neurobiological endophenotype for schizophrenia. First, the newer atypical antipsychotics are not yet available in Palau, and both unmedicated patients and those with remitted illness are available for evaluation. In Palau, schizophrenia patients may decline medication or elect to discontinue medication as long as they pose no threat to themselves or the community. Because Palauan families frequently live in remote areas, where relatives with schizophrenia can lead a relatively secluded life, it is common for individuals diagnosed with the illness to be unmedicated. Consequently, Palau offers a rare opportunity to study P50 sensory gating abnormalities in unmedicated schizophrenia patients, including patients with remitted symptoms and patients who have never received atypical neuroleptics. Second, these extended Palauan schizophrenia families, which combine multiple affected sibships linked by a common founder, have been drawn from a genetically isolated population
(40). Therefore, these extended families with their reduced genetic heterogeneity are well suited for subsequent linkage studies using the P50 sensory gating endophenotype.
In this study, medicated and unmedicated schizophrenia patients were compared to unaffected first-degree relatives and normal comparison subjects in order to determine whether the abnormal P50 sensory gating in schizophrenia patients and their relatives extends to this isolated non-Caucasian population. The objective was to evaluate the ability of the P50 sensory gating trait to serve as an endophenotype in a genetic linkage study of Palauan schizophrenia families.
Method
Subjects
A total of 85 patients diagnosed with narrowly defined schizophrenia and 83 of their first-degree relatives (46 parents and 37 siblings) were compared to 29 normal comparison subjects. In the schizophrenia patient group, 56 subjects were receiving therapeutic doses of typical antipsychotics (predominantly haloperidol and fluphenazine), and 29 subjects had been free of antipsychotic medication for at least 10 weeks but were clinically stable at the time of testing. Among the 29 unmedicated patients were 14 patients who were considered by the attending psychiatrist to be in full remission; that is, these patients had no clinically significant residual symptoms. For each schizophrenia patient who agreed to participate in a P50 sensory gating recording session, the biological parents and siblings were invited to participate in a similar recording session.
The patients and relatives were diagnosed according to a modified Schedule for Affective Disorders and Schizophrenia—Lifetime Version
(41), administered by a U.S.-trained, board-certified psychiatrist, supplemented by a review of psychiatric medical records. Consensual diagnoses based on the Research Diagnostic Criteria
(42) were established by an expert diagnostic panel, as reported previously
(40). Only patients with narrowly defined schizophrenia or schizoaffective disorder (with a mainly schizophrenic course) were included in the present study. Relatives diagnosed with an axis I DSM-III-R disorder were excluded.
The normal comparison subjects were recruited from families identified as unaffected during the ascertainment phase of the epidemiological study. To qualify, the subjects were required to be free of chronic psychiatric and neurological diseases and have no first-, second-, or third-degree relatives with psychotic illness. The latter qualification was fulfilled by using the Family Interview for Genetic Studies (http://zork.wustl.edu/nimh/figs/FIGS.pdf) to interview the subject and at least one other family informant. Because only a small proportion of the large multigenerational families found in Palau are exempt from mental illness and there are complex interconnections among families through marriage, this screening process yielded only 29 unrelated subjects.
Subjects showing evidence of chronic substance abuse were excluded. However, 76% of both male and female Palauans chew betel nut, which contains the alkaloid arecoline, and 63% chew betel nut with tobacco
(43,
44). As these usage levels suggest, betel nut is an integral part of daily life in Palau, just as caffeine is in other regions of the world. Therefore, betel nut users were not excluded from the present study, but they were required to abstain from betel nut chewing for 1 hour before the recording to control for the transient normalization of P50 sensory gating deficits that has been shown to follow nicotine administration
(45,
46).
In order to recruit as many qualifying subjects in each category as possible, we made no attempt to match the subjects in each group for age or gender. Among Palauan schizophrenia patients, the male-to-female ratio was previously reported to be 2:1, and the onset of illness is earlier for males than for females
(40). These epidemiological characteristics of the illness in Palau were reflected in a higher proportion of male subjects in the total patient group (72.9%, N=62) than in the normal group (37.9%, N=11) and a lower mean age for the patients (40.0 years, SD=10.4) than for the normal subjects (44.1 years, SD=17.4).
The study procedures were approved by the institutional review boards of the University of Utah and the Republic of Palau. All subjects provided written informed consent after receiving an explanation of the study in both English and Palauan from a Palauan coinvestigator.
Measurement of P50 Sensory Gating
Sensory gating was evaluated by recording the P50 wave of the auditory evoked response in a paired-stimulus or conditioning-testing paradigm, measuring the P50 wave amplitudes, and then calculating the P50 ratio, the ratio of the amplitude during testing to the amplitude during conditioning; lower values indicate better sensory gating
(47,
48). EEG activity was recorded by using gold electrodes affixed with electrode paste. The vertex (Cz) electrode was referenced to linked earlobe electrodes, and the ground was placed at the middle of the forehead. Electro-oculographic (EOG) activity was simultaneously monitored by using gold electrodes positioned at the outer canthus and below the right eye. Electrode impedance was less than 5 kW at all electrode sites.
The EEG and EOG data were collected by using Grass P511 amplifiers (Astro-Med, West Warwick, R.I.) set to an amplification of 50,000 with 1–300-Hz bandpass filters. The data were sampled at 1000 Hz by using custom-designed data acquisition programs written for a Windows 98 personal computer application (“NiftyP50”) and a 12-bit analog-to-digital converter. Calibration pulses were recorded for each session so that analog-to-digital resolution for the EEG and EOG channels could be calculated.
All recordings were made with the subject lying flat with his or her head and neck supported by a pillow, eyes open, and relaxed but awake. Clicks were delivered binaurally by using an auditory stimulator and insulated headphones. The click stimulus intensity was set to 50 dB above the subject’s threshold to optimize the evoked potential response while minimizing startle. The clicks were presented in pairs separated by 500 msec. The intertrial interval was randomly varied between 6.5 and 10.0 seconds (average=8.2 seconds). The click pairs were presented in three blocks of 24 conditioning-test trials, for a total recording time of approximately 12 minutes. The signals were measured between 100 msec prestimulus and 400 msec poststimulus.
Digitized data for each subject were stored in a database for subsequent analysis by a custom-designed analysis program written in C++. The data were screened and analyzed by raters blind to age, gender, and diagnostic status. Conditioning trials with no discernible positive wave in the 100-msec poststimulus interval and trials with evidence of EOG activity (e.g., eye blinking), myogenic artifacts greater than 40 mV, or alpha wave activity during the recorded epoch were rejected. For all nonexcluded trials in a recording session, the individual raw data traces were averaged within each sampling category (EEG and EOG for stimulus 1 versus stimulus 2), yielding four mean traces. Each of the four mean traces was digitally filtered in the frequency domain by using a fast Fourier transform (FFT) algorithm. Two FFT-based digital filters were applied in series: first, a low-pass filter that attenuates at 100 Hz, and second, a high-pass filter that attenuates at 30 Hz.
An algorithm was used to identify and quantify the P50 component of the filtered mean EEG traces. The P50 wave selected to represent conditioning was the most positive peak between 40 and 75 msec following stimulus 1. The P50 peak representing the test condition was then selected as the most positive wave during a latency range equal to the latency of the conditioning response plus or minus 5 msec. Amplitude was defined as the difference between the positive peak and the preceding negative trough for both the conditioning P50 wave and the test P50 wave. The P50 ratio was calculated as the test P50 amplitude divided by the conditioning P50 amplitude. This P50 ratio is thus a measure of the gating, or inhibition, of the P50 response, and lower values indicate greater auditory sensory gating.
Results
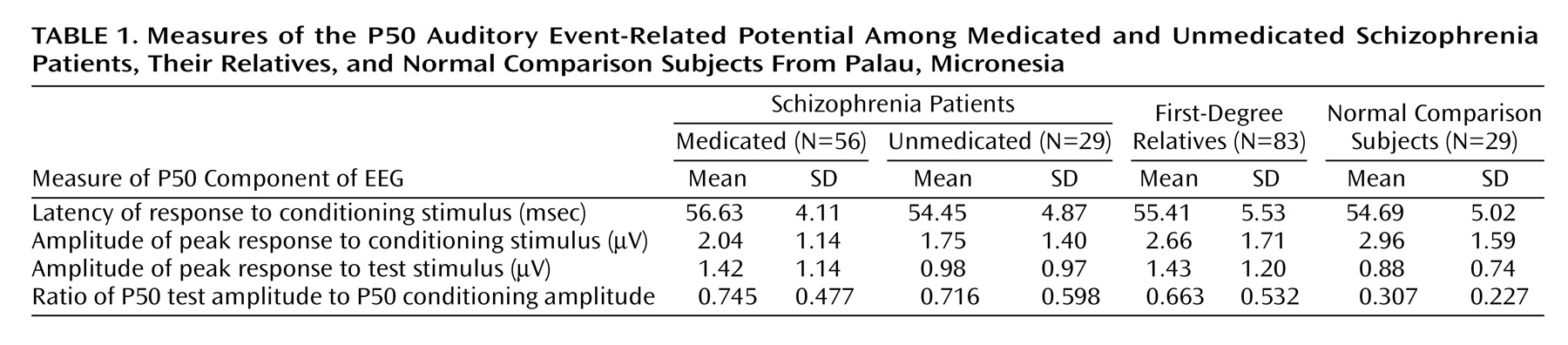
Table 1 presents the mean values and standard deviations for each of the event-related potential measures (conditioning latency, conditioning amplitude, test amplitude, and P50 ratio) for the medicated and unmedicated schizophrenia patient groups, the relatives, and the normal comparison subjects. To determine the presence of group differences in P50 sensory gating, we conducted analyses of variance on each of the measures presented in
Table 1.
Auditory sensory gating as represented by the P50 ratio was impaired in both the medicated and unmedicated schizophrenia patients and in their first-degree relatives, compared to the values for the normal comparison subjects (F=4.85, df=3, 189, p=0.003). The mean P50 ratios were equivalent across the patient and relative groups according to Tukey’s comparison test (p<0.05).
There were significant group differences in the P50 conditioning amplitude (F=3.20, df=3, 189, p=0.03) and test amplitude (F=2.67, df=3, 189, p=0.05) but not in conditioning latency (F=2.06, df=3, 189, p=0.11). Tukey’s test revealed that both patient groups had lower P50 conditioning amplitudes than the normal subjects (p<0.05). The relatives and the medicated patients showed larger responses to the second stimulus than did the normal subjects, as reflected in the P50 test amplitudes.
For the medicated patients, Pearson correlations showed no significant effect of medication dose (measured in chlorpromazine-equivalent units) on any of the P50 variables. None of the event-related potential measures showed a significant main effect of gender or gender-by-group interaction, and age was nonsignificant as a covariate.
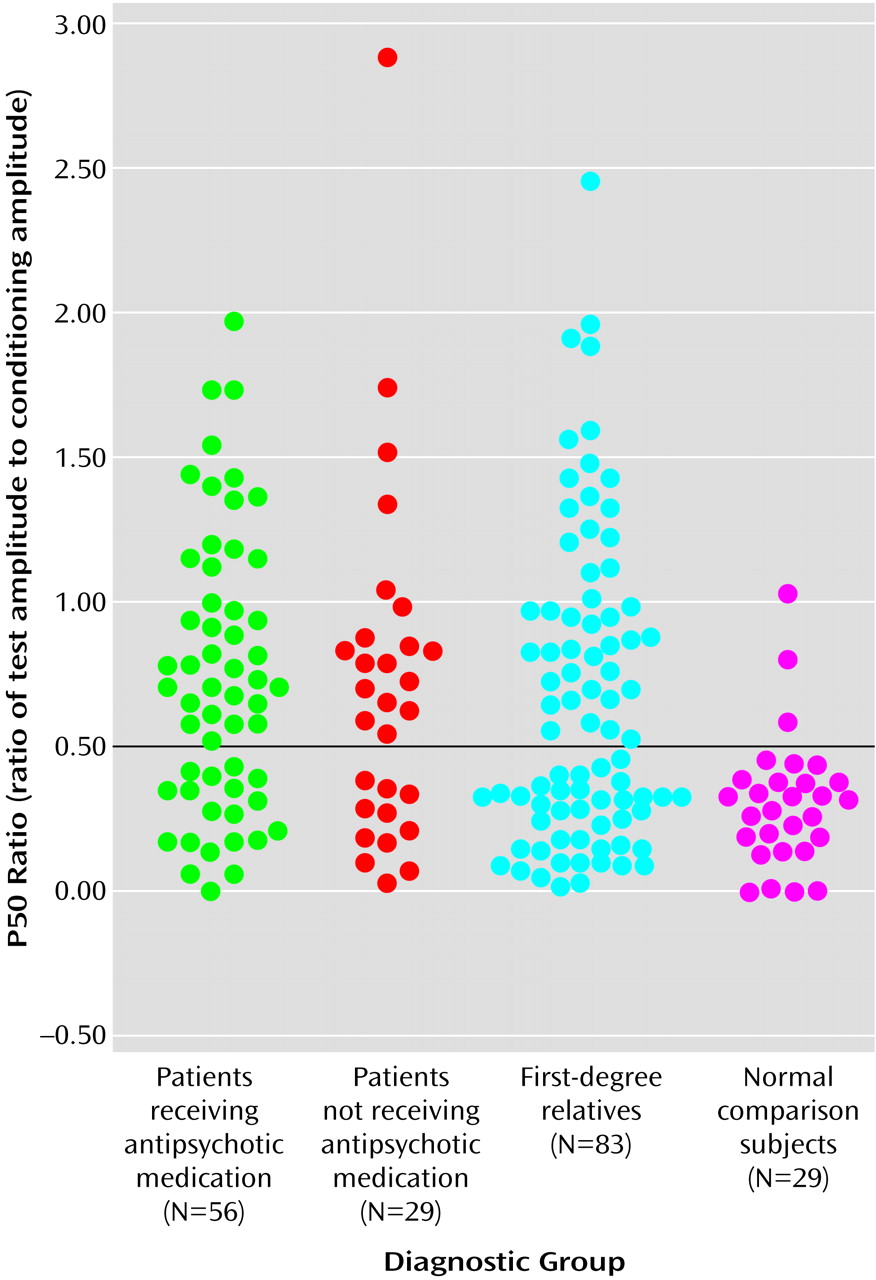
Figure 1, which presents the distribution of P50 ratios for the two patient groups, the relatives, and normal comparison subjects, indicates that P50 ratios of 0.50 and above were rare in the comparison group, occurring in only three (10.3%) of the 29 normal subjects. This 0.50 threshold value for the P50 ratio is the same threshold used by Freedman et al.
(48) in a linkage study of Caucasian families with schizophrenia, indicating that the fundamental characteristics of the auditory event-related potential are similar in these Pacific Islanders. When P50 ratios above 0.50 were designated as “abnormal,” the P50 sensory gating deficit occurred in 37 (66.1%) of the 56 medicated patients, 18 (62.1%) of the 29 unmedicated patients, and 43 (51.8%) of the 83 relatives. Because the rate of abnormal gating did not differ between the two patient groups (χ
2=0.13, df=1, n.s.), they were combined. Chi-square tests confirmed that the rates of abnormal gating were significantly higher in both the combined patient group (χ
2=25.57, df=1, p<0.001) and the first-degree relatives (χ
2=15.27, df=1, p<0.001) than in the normal comparison group. The resulting relative risk for abnormal P50 sensory gating is 5.0 (calculated by dividing 51.8%, the “affection” rate in relatives, by 10.3%, the “affection” rate in normal subjects).
Discussion
The objectives of our study were to determine whether abnormal P50 sensory gating in schizophrenia families extends to this Pacific Islander population and, by examining the effects of medication status on P50 sensory gating performance, to evaluate the ability of the P50 inhibitory trait to serve as an endophenotype in a genetic linkage study of these families. P50 sensory gating deficits that have been found in Caucasian schizophrenia patients and relatives were confirmed in this isolated Pacific Island study group. This confirmation of a similar pattern of deficits in non-Caucasian subjects is important because schizophrenia is phenomenologically similar worldwide and the underlying neurobiological disruptions associated with the illness should apply across ethnic groups.
Our findings confirm the presence of impaired auditory sensory gating in both medicated and unmedicated schizophrenia patients. Patients who had been free of typical antipsychotic medication for at least 10 weeks were just as likely to exhibit the P50 sensory gating deficit as patients who were receiving therapeutic doses of typical antipsychotics. Furthermore, in the medicated patient group, the P50 sensory gating ratio was uncorrelated with medication dose. This rare opportunity to study schizophrenia patients in a naturalistic setting revealed that impaired P50 sensory gating occurs independently of the effects of typical antipsychotics in a variety of patients at different stages of the illness. These results suggest that in Palauan subjects, the P50 paradigm measures a stable neurobiological trait that does not fluctuate with clinical state or treatment, an important characteristic for a trait marker being considered as an endophenotype in genetic studies of schizophrenia. Also, age and gender effects on P50 sensory gating performance were nonsignificant in these Palauan subjects.
Impaired P50 sensory gating, defined in the present study as a P50 ratio greater than 0.50, was present in only 10% of normal subjects but in 65% of schizophrenia patients and over 50% of their first-degree relatives. Therefore, the relative risk for abnormal P50 sensory gating is 5.0, solid evidence for the potential value of using the P50 ratio as an endophenotype in a genetic linkage study of these Palauan schizophrenia families.
A linkage study conducted in multiplex schizophrenia families of European ancestry
(48) provided significant evidence for linkage of the P50 sensory gating phenotype to a chromosome 15q13-14 locus containing the alpha-7 nicotinic receptor. At the present time, this group of Palau families may be the only other existing set of families with sufficient data on P50 sensory gating to test for replication of this result. This genetic linkage study, which is currently in progress, will first test for linkage of the qualitative P50 sensory gating endophenotype to the chromosome 15q13-14 locus identified by Freedman et al.
(48) and then examine the chromosome 2p13-14 region, where we have found significant evidence for linkage to the diagnostic phenotype in the Palau families
(49,
50).
Given the stability of the trait and the absence of gender and age effects in these Palauan subjects, the P50 ratio may be highly suitable for quantitative trait locus linkage analysis in addition to the more traditional linkage analysis approaches. Therefore, my colleagues and I intend to conduct a genome-wide analysis of linkage with the quantitative P50 sensory gating data in these Palauan families using recently developed oligogenic linkage analysis methods
(51,
52) that can detect quantitative trait loci influencing endophenotypic variation. This more comprehensive linkage approach may be able to detect additional chromosomal regions implicated in the P50 sensory gating deficit and ultimately identify susceptibility loci that are more directly relevant to the functional impairments that characterize schizophrenia.